Article Text

Statistics from Altmetric.com
Description
A 57-year-old man presented with a 6-month history of recurrent epistaxis and melena. At presentation, he was haemodynamically stable and laboratory findings revealed iron deficiency anaemia (Hb=9.9 g/L, MCV=77 fL) with low ferritin (88 ng/mL).
A gastrointestinal cause of anaemia was suspected and oesophagogastroduodenoscopy (OGD) was performed, revealing multiple reddish lesions in the gastric fundus, body and antrum as well as in the duodenum consistent with telangiectasias, non-bleeding at the time of the examination (figure 1), treated endoscopically with argon plasma coagulation.

Oesophagogastroduodenoscopy (OGD) depicting a duodenal telangiectasia (white arrow).
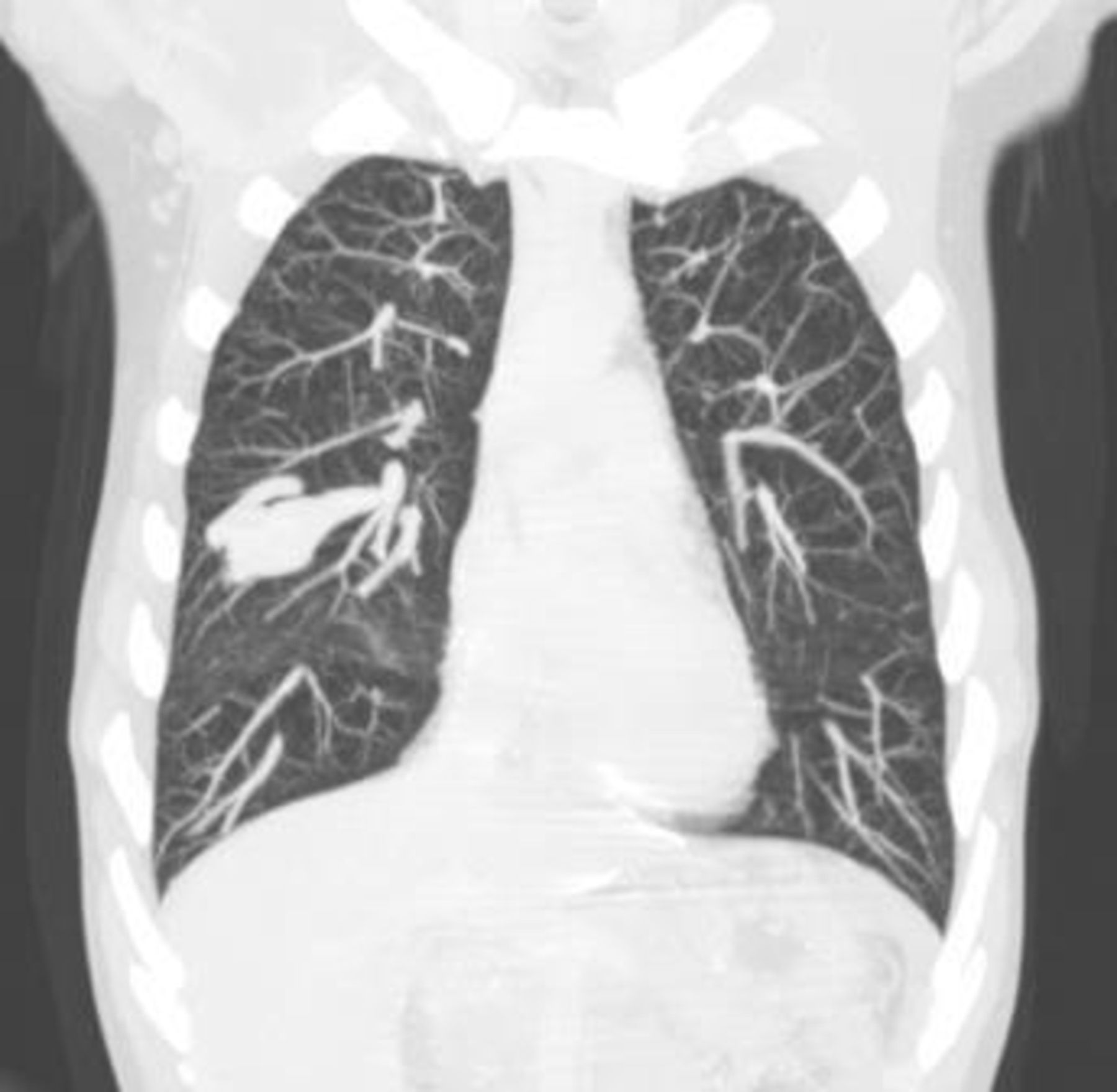
CT of the chest (figure 2) revealed a nodular lesion of parenchymatous density with well-defined edges, localised in the right middle lobe, measuring 45×23 mm. After intravenous administration of iodine contrast (figure 3), an arteriovenous malformation (AVM) was confirmed, with the afferent arterial vessel measuring 4 mm of diameter and a ectasic (7 mm) efferent vessel was identified originating from the AVM nidus and flowing into the upper right pulmonary vein. Based on the Curacao diagnostic criteria, the patient was diagnosed with hereditary haemorrhagic telangiectasia (HHT), also known as the eponym Osler-Weber-Rendu.1 Mutation of endoglin gene was confirmed in the patient, and genetic counselling was offered to the remainder family members (brother and sister - genetic testing currently underway).

Maximum intensity projection coronal view of a chest CT portraying a well-defined, elongated lesion, located in the right middle lobe.

{kind=link}
{kind=link}
{kind=link}
Maximum intensity projection axial view of a chest CT with iodine contrast illustrating the pulmonary arteriovenous malformation, with the central nidus (asterisk), the afferent arterial vessel (open white arrow) and efferent venous vessel (solid white arrow).
First-line treatment with bevacizumab and second line treatment both with tamoxifen and thalidomide was performed, but the patient remained with epistaxis, GI blood loss and anaemia requiring chronic ferric carboxymaltose therapy.2
HHT is a rare autosomal dominant inherited disorder which may give rise to arteriovenous malformations characterised by formation of abnormal vascular structures in telangiectasic shape in the microcircle of the derm, frequently involving the various mucosal surfaces, gastrointestinal tract and distal portion of the limbs.3
Vascular involvement of other organs occurs in an important percentage of patients, with cerebral vascular malformations (CVM) affecting 23% of diagnosed patients with HHT, and patients can present with stroke and brain abscess because of paradoxical emboli. Liver vascular malformations (LVM) are present in 32%–78% of patients but only a minority (8%) of those presenting with related symptoms, such as high-output heart failure, portal hypertension and biliary necrosis.1 The patient was submitted to transthoracic echocardiography (without contrast administration) and brain MRI with angiographic study, with no abnormal findings reported.
Pulmonary arteriovenous malformations (PAVM) occur in 15%–50% of cases, and have been associated with life-threatening and debilitating complications such as massive haemoptysis, spontaneous haemothorax, stroke, transient ischaemic attack and cerebral abscess.1
The size of the feeding afferent arterial vessel (more than 3 mm in diameter) is used as a criterion for treatment of PAVM, regardless of the presence or absence of symptoms. Angiographic occlusion with embolisation has been shown to be efficacious and have a good safety profile and is the treatment of choice. Thoracic follow-up should be done with chest CT within 6–12 months following embolisation and at 3-year intervals thereafter is recommended.1
Learning points
Haemorrhagic telangiectasia (HHT) is a rare autosomal dominant involving the various mucosal surfaces and with visceral vascular disease in an important percentage of patients.
Pulmonary AVM’s can be associated with life-threatening and debilitating complications and the angiographic occlusion with embolisation is the treatment of choice.
Footnotes
Contributors JA wrote the manuscript. CF performed the radiological studies. CLV performed the endoscopic studies, treated and followed the patient. CF and CLV revised the manuscript.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Patient consent Obtained.
Provenance and peer review Not commissioned; externally peer reviewed.