Article Text

Summary
McCune-Albright syndrome (MAS) is a rare disease characterised by triad of monostotic or polyostotic fibrous dysplasia, café au-lait skin spots and a variety of endocrine disorders; precocious puberty (PP) being the most common presenting symptom in female patients. Hyperfunction endocrinopathies including hyperthyroidism, growth hormone excess and cortisol excess are typical presentations in MAS. We present a case of 21-year-old woman with clinical and radiological characteristics of MAS triad; she presented with short stature which was attributed to both growth hormone deficiency and PP. Growth hormone deficiency in MAS has not been reported in English medical literature.
- endocrine system
- skin
- osteoporosis
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
McCune-Albright syndrome (MAS) is a rare genetic disorder that was first described in 1936 by Dr Donovan McCune and Dr Fuller Albright. MAS affect 1 in 100 000 to 1 in 1 000 000 people worldwide. It involves a triad of poly/monostotic fibrous dysplasia (FD), café-au-lait spots and endocrinopathies (predominantly involving precocious puberty in girls).1–5 Other common endocrinopathies involved in MAS include: hyperthyroidism, growth hormone (GH) excess, hyperprolactenaemia, renal phosphate wasting, Cushing syndrome and ovarian cysts.2 6 7 Several other organ systems such as liver, heart, parathyroid and pancreas may occasionally be involved.8
MAS result from activating somatic mutations of the GNAS1 gene on the long arm of chromosome 20; specifically affecting Gsα, a cAMP regulating protein. The mutation occurs during embryonic development and extent of disease and tissues involved are determined by the stage of embryonic development and survival of mutated cells. Activation of Gsα is responsible for varied nature of endocrinopathies that occur in MAS.1 5–7 Gsα also acts as an ‘on and off’ switch protein to other body areas including skin (café au lait), ovary (precocious puberty) and bone (FD).1 Hypophosphataemia in MAS is explained by excess fibroblast growth factor 23 (FGF-23), a hormone produced by fibrous dysplastic bones and it causes the kidneys to lose phosphorus in the urine.1
Our index case is peculiar because at presentation and even during follow-up, there was persistence of GH deficiency.
Case presentation
A 21-year-old woman presented at the endocrinology clinic complaining of short stature compared with her siblings and peers. She had a medical history of precocious puberty; her mother narrated that irregular menses started at the age of 3 years and only become regular when she was 9 years. Patients’ breasts were also reported to develop since early childhood however galactorrhoea was not observed. There was no history of bone fractures. The rest of patient and social history was not significant. Physical examination revealed a young woman with short stature: height: 1.37 m, weight: 37 kg, temperature: 36.7°C, pulse rate: 86 beats per minute, blood pressure: 115/68 mm Hg, café-au-lait skin spots were found on the middle line of the back (figure 1) and lower abdomen. Both breasts were well developed (Tarner V), with adequate pubic hair distribution (Tarner IV), normal external female genitalia and normal clitoris. Visual fields and hearing were normal.

Café-au-lait spots in the middle line of the back.
Investigations
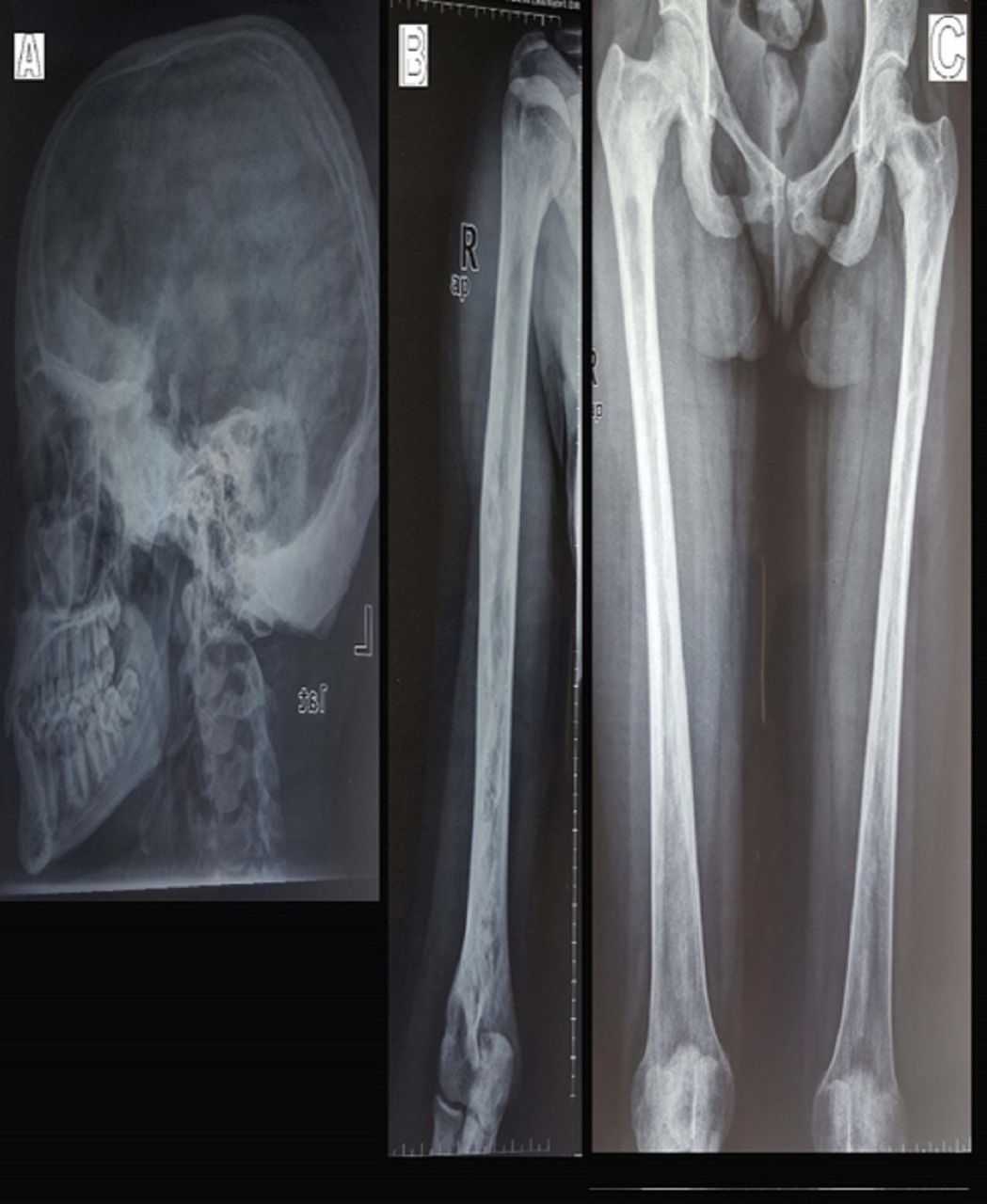
Routine laboratory tests and serum levels of luteinising hormone (LH), follicle-stimulating hormone (FSH), dehydroepiandrosterone sulfate (DHEAS), estradiol, prolactin, thyroid-stimulating hormone (TSH) and T4, adrenocorticotrophic hormone (ACTH) and basal cortisol were all within normal ranges. Unexpectedly serum insulin-like growth factor 1 (IGF-1) level was low (68 ng/mL, N: 267.5–470.8); subsequent clonidine test was done showing a GH deficiency with serum GH: 0.43 ng/mL, 0.29, 0.13, 0.09 and 0.08 at 0, 30, 60, 90 and 120 min (<5 ng/mL indicative of GH deficiency). Abdominal-pelvic ultrasound revealed normal liver, gall bladder, kidneys, spleen and pancreas. Uterus, endometrium and left ovary appeared normal, whereas a follicular cyst measuring 2.7 cm in diameter was noted in the right ovary. Ultrasound of the neck ruled out thyroid and parathyroid glands enlargement. The brain MRI showed T1 hypointense signal lesions with bone expansion, remodelling and cranial asymmetry involving the frontal, parietal, occipital, sphenoid, clivus, right petrous-temporal and zygoma and floor of the sella turcica, with fullness of sphenoid sinus suggestive of FD (figure 2). FD was confirmed with a bone biopsy in histopathology department of hospital abroad. X-ray bone survey showed polyostotic FD involving skull, humeri, both femur and tibia (figure 3). Genetic testing was not available in our setting. The diagnosis of MAS was made based on clinical and radiological findings.
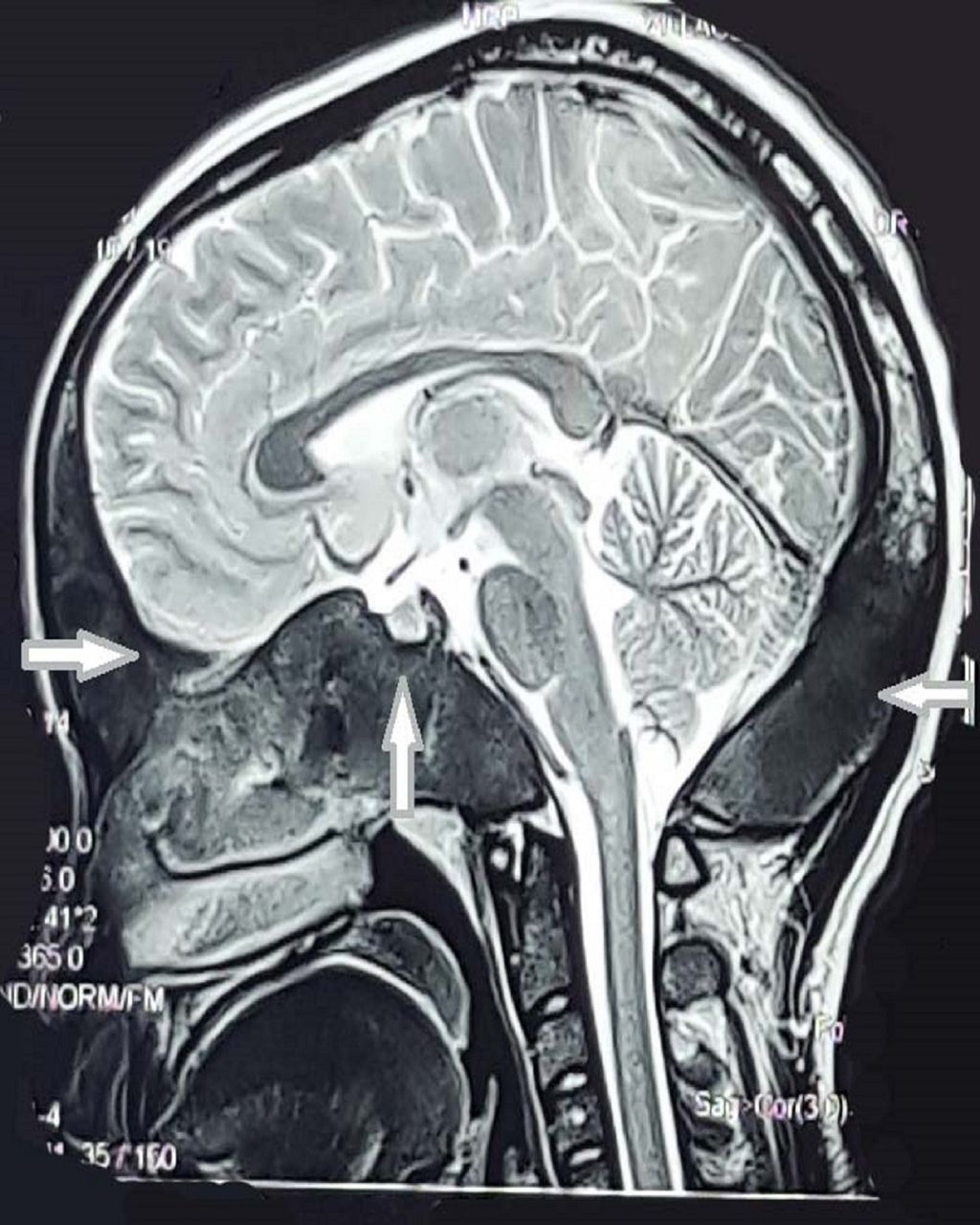
Brain MRI shows bone expansion and remodelling involving the frontal, parietal, occipital, sphenoid, clivus, right petrous-temporal, right zigoma and floor of sella turcica with fullness of sphenoid sinus.
{kind=link}
{kind=link}
{kind=link}
(A) Skull: sclerosis and expansion of the frontal bone and floor of the anterior cranial fossa extending into the sphenoid bone. Marked thickening and sclerosis of the occipital bone and posterior parietal bone. (B) Humerus: thickening of the cortex of the diaphysis, cystic lesions with thickened sclerotic rind in the methadiaphysis. (C) Femur: patchy sclerosis and expansion of the neck of both right and left femur, cortical thickening, scalloping and ground glass with radiolucency in the diaphysis bilaterally.
Treatment
The patient was treated with bisphosphonates in the form of ibandronate at a dosage of 150 mg monthly to reduce the risk of pathological fractures for over the past 1 year of follow-up.
Outcome and follow-up
The patient has been followed up at endocrinology clinic for over the past 1 year. There is no change of clinical or laboratory sequela with recent hormone profile revealing persistence of GH deficiency (IGF-1=128 ng/mL; GH at rest=0.07 ug/L). Other hormone profiles (LH, FSH, DHEAS, estradiol, prolactin, TSH and T4, ACTH and basal cortisol) have remained within the normal ranges. Haemogram, serum electrolytes, renal functions and liver enzymes are within the normal ranges except for raised alkaline phosphatase (ALP)=154 μmol/L (35–110). Bone profile results were as follows: corrected serum calcium=2.31 mmol/L (2.10–2.55); serum phosphate=0.69 mmol/L (0.87–1.47); 24-hour urine calcium and phosphate were 1.48 mmol (0.8–1.8) and 14.93 mmol/L (12.00–42.00), respectively; vitamin D25-OH=32.70 ng/mL (>20 indicate sufficiency); PTH=60 pg/mL (15.0–65.0). Tests for vitamin D (1, 25(OH) 2D) and FGF-23 were not performed as they are not done both in the country and in Republic of South Africa.
Discussion
MAS is always diagnosed on the basis of clinical background, supported by radiological findings of FD. Though the disease is caused by somatic mutation that occurs during embryonic stage, genetic testing is not necessary and there is no indication to screen other family members as the disease is not inherited.5 7
Short stature attributed to both GH deficiency and precocious puberty has not been reported in the triad of MAS; however, isolated cases of FD have reported the mechanism with which short stature may occur. Premature fusion of epiphysial growth plates secondary to defects in osteoblastic differentiation and maturation, hence resulting in short stature9 10; we speculate this is one possible mechanism of short stature in our patient as compared with her siblings and peers. On the other hand, GH deficiency found persistently in our patient could as well have contributed her phenotype. Oestrogen excess occurring early when patient had precocious puberty might also have accelerated bone maturation and short stature.11 There was no documented follow-up of height for us to elicit this possibility.
Our patient presented with severe FD involving several bones in the scalp; we believe that GH deficiency most likely resulted from compression of pituitary region responsible for GH production by expanded dysplastic bones.
Despite the fact that our patient presented with widespread FD involving multiple bones; she did not present with bone pains and deformities; asymptomatic FD without bone pains or deformity has been described previously.12 Furthermore, diagnosis of FD in our patient was made through radiological findings after patient’s clinical evaluation revealed the other two components of the triad. Findings on radiological images included sclerosis and expansion of several skull bones, cystic lesions and ground-glass appearance with radiolucency; these are typical features as described by other authors.13
Hypophosphataemia in FD/MAS is usually caused by elevated levels of FGF-23.14 FGF-23 downregulates expression of sodium–phosphate cotransporters in the proximal tubule to stimulate phosphaturia resulting in hypophosphataemia. On the other hand, FGF-23 stimulates 24-hydroxylase to suppress the production of 1,25 dihydroxyvitamin D (1, 25-(OH) 2D).15 16 Normal PTH rules out a possibility of hyperparathyroidism-stimulating bone secretion of FGF-23.17
Analysis of the biochemical bone profile of our patient revealed mild hypophosphataemia and normal 24-hour urinary phosphate. Low dietary phosphate is one of the possibilities that can explain these findings. High dietary phosphate intake is known to cause high levels of FGF-23, while low dietary phosphate could cause the opposite resulting to hypophosphataemia and normal urine phosphate.18 19 Since we could not identify the phosphate content in the diet of our patient, our reasoning remain a speculation. Second explanation for not having elevated phosphate in urine is the fact that our patient had advanced bone disease which is associated with less bone activity, hence, less FGF-2320 21; this cannot be confirmed as this test is not available in our setting. On the other hand, postzygotic mutations of GNAS1 gene have been shown to account to lack of renal phosphate wasting in about 50% of patients with MAS/FD.22 Genetic testing was not performed in this case.
The rarity of MAS coupled with different manifestations of the condition depending on tissue involved makes choice of any specific treatment to be difficult; hence, management of the patient is based on controlling symptoms, strengthening exercises, stabilising the skeleton to prevent fractures and providing bisphosphonates which have been shown to help in reducing bone pains without necessarily altering natural history of the disease.7 23–25 Our patient has been receiving bisphosphonates in the form of ibandronate for over the past 1 year and she has not had any bone pains or fractures.
Despite persistence of GH deficiency, we did not prescribe GH analogues to our patient because the latter has been shown to make craniofacial FD worse.26 27
Learning points
McCune-Albright syndrome (MAS) presenting with a range of skeletal and extraskeletal manifestations requires extensive clinical, laboratory and radiological work up to enhance proper identification of extent of tissues involved.
The case highlights the importance of appreciating the fact that hypofunction endocrinopathies manifestations may be a reason that patients seek medical attention.
Short stature in MAS even in patients with a history of precocious puberty should alert physicians to seek for possible underlying growth hormone deficiency.
Acknowledgments
The authors would also like to acknowledge the fact that the abstract for this case report was first submitted and accepted under the title ‘McCune- Albright Syndrome with Growth Hormone Deficiency: Case report’ and published (not presented) in conference proceedings of the 99th Annual Meeting of the Endocrine Society 2017 (1–4 April 2017). It is available online at: https://endo.confex.com/endo/2017endo/meetingapp.cgi/Paper/30124.
References
Footnotes
Contributors YPR and GMR: wrote the first draft of the manuscript; interpreted the clinical and laboratory findings. SS: interpreted the radiology images. All the authors read, corrected and approved the final manuscript.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Patient consent Obtained.
Provenance and peer review Not commissioned; externally peer reviewed.