Article Text

Statistics from Altmetric.com
Exertional dyspnoea is commonly an early feature in respiratory disease; however, neurological disease may limit mobility and, as a consequence, preclude this symptom. Diagnosis of respiratory dysfunction resulting from neurological disease may therefore require a higher index of clinical suspicion or the application of specific tests; this exercise is worthwhile if it allows advance detection and discussion and (where appropriate) treatment, of impending overt respiratory dysfunction. Specific symptoms and appropriate tests will be discussed in the text and have also been reviewed in detail elsewhere.1 However, it should be recalled that, at the most basic level, the function of the respiratory muscle pump is to produce inspiratory airflow, which is related to the ability to generate a subatmospheric pressure within the thorax. Thus, although access to detailed investigation of respiratory muscle is not universal, we encourage measurement of both the lying and standing vital capacity2 and static mouth/nasal pressures,3 4 which can be done either in the neurological clinic or in any standard lung function laboratory.
This review deals with acute neuromuscular respiratory disease (including those aspects of respiratory muscle function relevant to intensive care), chronic neuromuscular respiratory disease, sleep related disorders, respiratory consequences of neurological disease, and finally with neurological features of respiratory disease.
Acute neuromuscular respiratory disease
The presentation of acute ventilatory failure due to neurological disease may be genuinely acute or may simply result from any of the causes of chronic respiratory neuromuscular dysfunction, which are discussed below, passing undiagnosed. Patients who present with acute ventilatory failure and no diagnosis usually receive treatment in the form of mechanical ventilation before a diagnosis is reached. The cause may be disease of the nerves, the neuromuscular junction, or muscle5; however, most data relating to the assessment of such patients have been obtained from the study of patients with Guillain-Barré syndrome, which is dealt with below. Various other causes, particularly heavy metals, may occasionally cause respiratory failure due to nerve damage (table).6 Acute poliomyelitis is seldom encountered in the western world but still occurs in the developing world.7
Neurological disorders that can result in respiratory failure
NERVE
The acute motor neuropathy of Guillain-Barré syndrome and its variants commonly involves the nerves supplying the respiratory muscles, leading to the need for mechanical ventilation in 14% of cases.6 It is now established that Guillain-Barré syndrome may be effectively treated with either plasmapharesis or intravenous immunoglobulins. However, despite these treatments, the mortality and residual morbidity remain significant even in the most experienced hands. Ng et al treated 80% of their series using one or other of these therapies; nevertheless, the mortality of patients requiring admission to an intensive care unit was 5.1%.8 Frequent vital capacity measurements are mandatory as a fall in vital capacity precedes the requirement for mechanical ventilation, which on average occurs when the vital capacity falls below 15 ml/kg body weight.9 Other reported predictors of the need for ventilatory support include cranial nerve involvement, the history of an infection in the 8 days before the onset of Guillain-Barré syndrome, and a greatly increased CSF protein.10 A reduction in the amplitude of the action potential recorded from needle electrodes sited in the diaphragm after phrenic nerve stimulation is associated, statistically, with the need for mechanical ventilation.11 However, in this study abnormal responses were also found in 79% of patients who did not subsequently require mechanical ventilation; thus needle EMG of the diaphragm does not provide clinically useful information unless the results are entirely normal, in which case ventilation is unlikely to be required. Non-invasive mechanical ventilation has not, to the best of our knowledge, been described in and is not, in our view indicated, given that bulbar dysfunction is a recognised feature of the condition.8 Weaning from endotracheal ventilation can be difficult or even impossible. Prognostic factors indicating a greater eventual disability are need for and duration of ventilatory support, age,8 12 and the action potential amplitude obtained by stimulation of peripheral nerves.12 13 Two large recent studies have compared weaning methods14 15 and reached differing conclusions. Patients with neurological disease comprised less than 20% of the subjects in both studies and therefore the optimal weaning method in Guillain-Barré syndrome is still unknown.
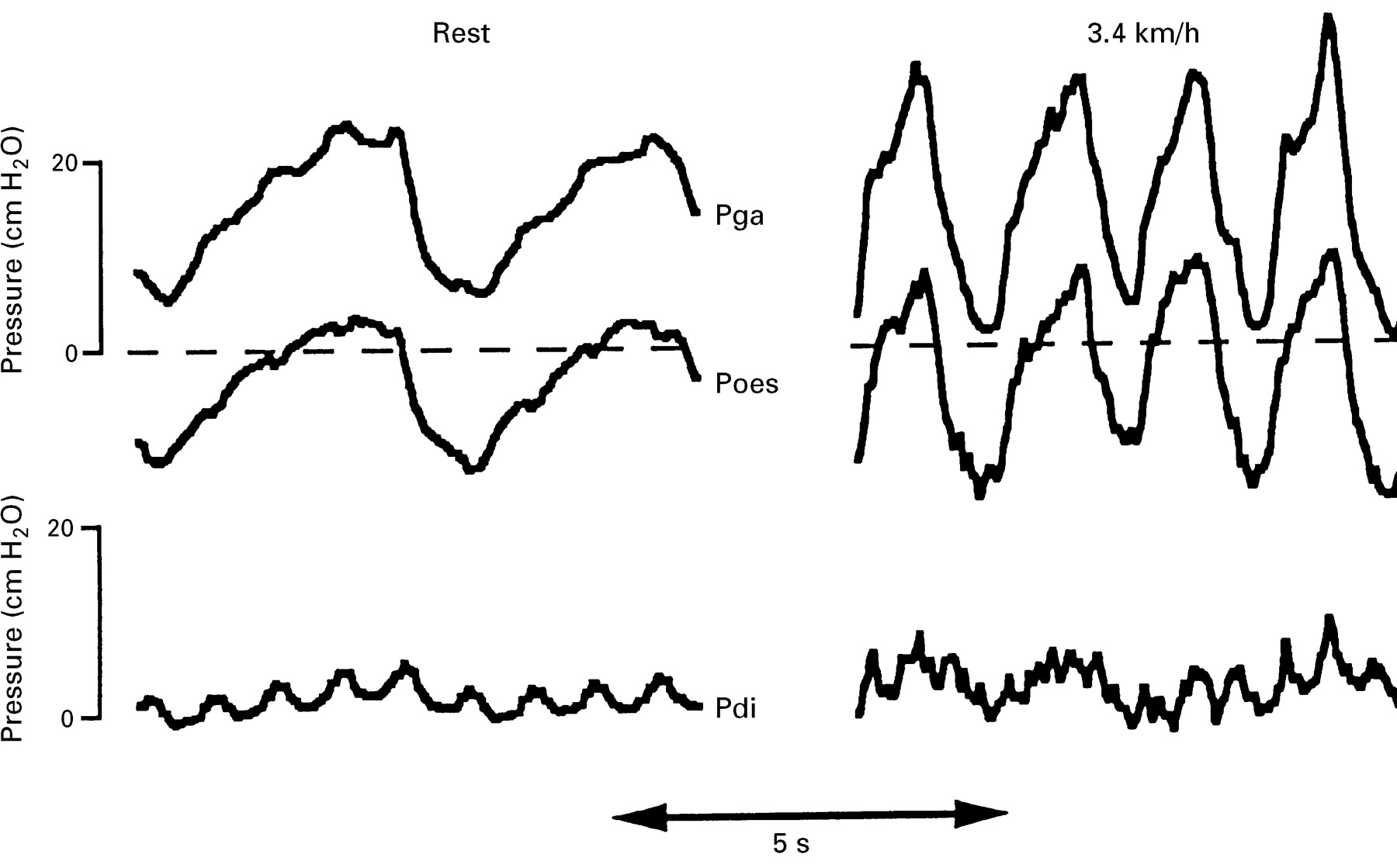
We are not aware of any studies considering the relative contributions of the different muscle groups which constitute the respiratory muscle pump to the ventilatory failure of Guillain-Barré syndrome. The abdominal muscles are, clinically, often involved in Guillain-Barré syndrome; these muscles are the principal muscles both of active expiration16 and of cough.17 Abdominal muscles are recruited at low levels of ventilatory activity in normal subjects.18 Their importance may be judged from the fact that comparatively high levels of ventilation may be achieved by predominant use of the expiatory muscles.19 Conversely, patients in whom the abdominal muscles are absent cannot generate an appropriate ventilatory response to exercise.20 A further example of the importance of the abdominal muscles may be seen when patients with isolated paralysis of the diaphragm exercise (fig 1). Unlike normal subjects, who may generate diaphragmatic pressure (Pdi) swings of more than 20 cm H2O,21 these subjects generate no Pdi but use their abdominal muscles (as shown by the expiratory rise in gastric pressure (Pga) to reduce end expiratory lung volume below functional residual capacity to facilitate the subsequent inspiration.
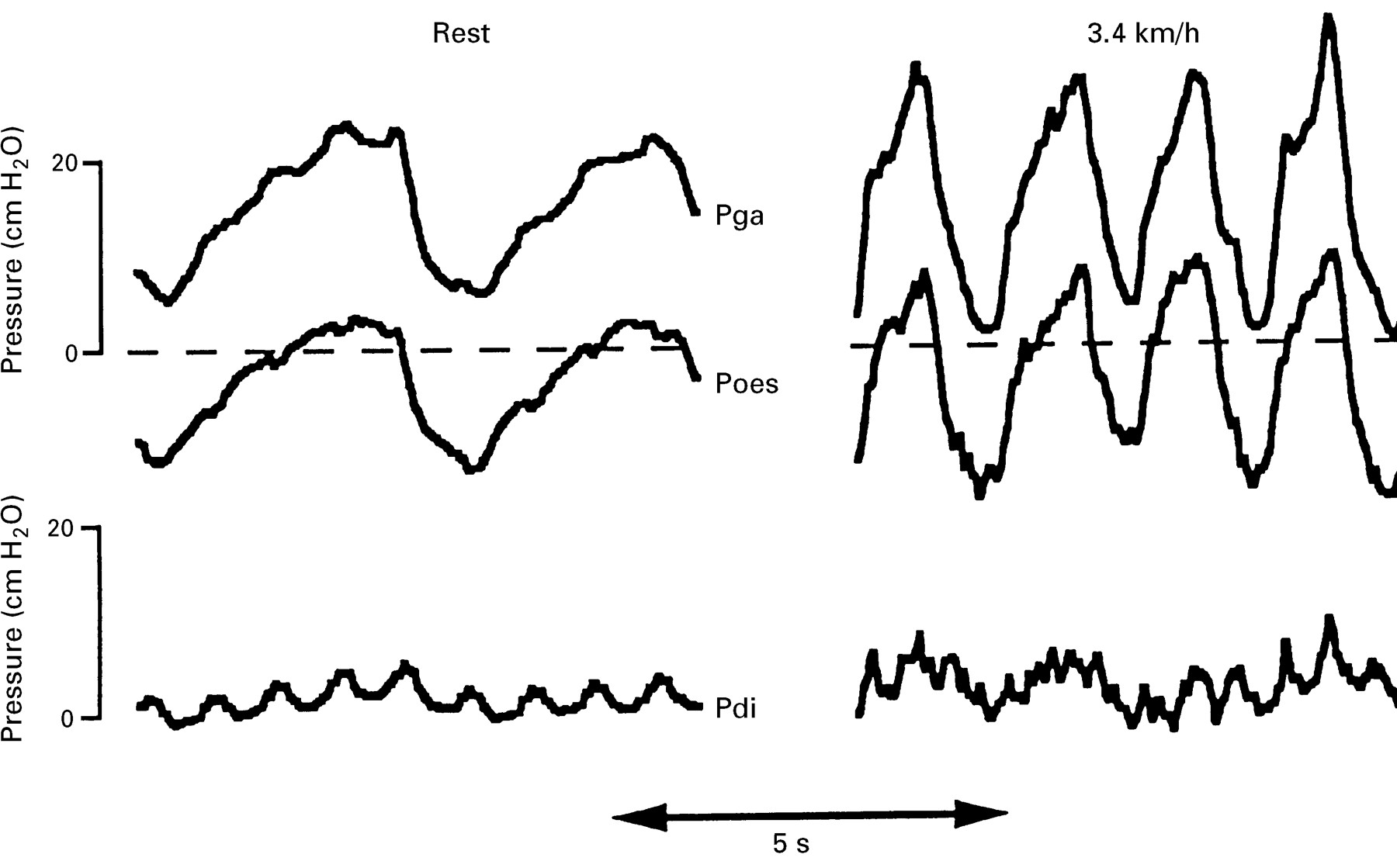
Recordings are shown from a patient with bilateral diaphragm paralysis due to neuralgic amyotrophy at rest (left panel) and walking on a treadmill at 3.4 kmh-1 (right panel). When exercising there is an increase in respiratory rate and recruitment of the abdominal muscles such that there is an increase in the peak (end expiratory) gastric pressure (Pga). This pressure is transmitted to the thorax, as shown by the oesophageal pressure (Poes). The Pdi is negligible in both conditions (lower traces), confirming diaphragm paresis. Abdominal muscle recruitment is also demonstrated by patients with an intact diaphragm but such patients would also develop Pdi swings (typically 20 cm H2O).
Assessment of respiratory muscle function in the intensive care unit
Measurements of inspiratory muscle strength on the intensive care unit should theoretically be of value in the prediction of weaning; however, such measurements are difficult to obtain reliably. In non-intubated patients the finding of a normal supine vital capacity is of value for excluding clinically relevant inspiratory muscle weakness.2 However, the vital capacity can be difficult to measure on the intensive care unit.22 The maximal inspiratory pressure has been proposed for use in Guillain-Barré syndrome23 but this test is, in general, not a good predictor of weaning success.24 Measurement of transdiaphragmatic pressure25 or sniff mouth pressure26 does not confer any additional advantage indicating that the likely reason that such tests are poor predictors of weaning outcome is because they rely on the assumption that the patient is able (and willing) to fully activate the diaphragm during a voluntary manoeuvre, which is not always valid.27 This may be circumvented by stimulating the nerves artificially using bilateral anterior electrical28 or magnetic29 phrenic nerve stimulation and measuring the transdiaphragmatic or endotracheal tube pressure.30 Preliminary data suggest that such measurements are possible both in adults31 and children.32 However, it remains unknown whether these non-volitional tests will be more effective predictors of weaning success.
Neurological complications of critical illness
Wasting is a relatively common clinical finding on the intensive care unit but respiratory or limb muscle weakness that is sufficiently severe to prompt formal investigation occurs in only 1% of intensive care unit admissions33; the weakness is acquired on the intensive care unit in 70% of these. Neuromuscular weakness acquired in the intensive care unit may be due to critical care myopathy or critical illness polyneuropathy; patients at increased risk are transplant organ recipients (who are also more likely to receive corticosteroids and neuromuscular blockers) and those with multiple organ dysfunction.33 Critical illness polyneuropathy is a complication of multiple organ failure which is thought to occur in up to 50% of such patients,34 and is characteristically associated with reduced amplitude of the diaphragmatic action potential with relative preservation of phrenic nerve latency.35 It may be difficult to differentiate this syndrome from the acute myopathy which has also been reported from patients in the intensive care unit,36 and the prognosis is similar.33Various drugs commonly used on the intensive care unit, including classic antiarrhythmic drugs, may induce respiratory neuromuscular blockade.37 No studies to date have attempted to assess the impact of prolonged intensive care unit admission on skeletal muscle strength, but preliminary data obtained by our group from studies in the quadriceps muscle using the technique of magnetic stimulation of the femoral nerve38 suggest that loss of tension generating capacity may be considerable39 and relatively slow to recover (fig 2). No specific therapy is available for neurological illness related to critical care, but there is increasing recognition that prolonged use of neuromuscular blockade should be avoided36 unless absolutely required.
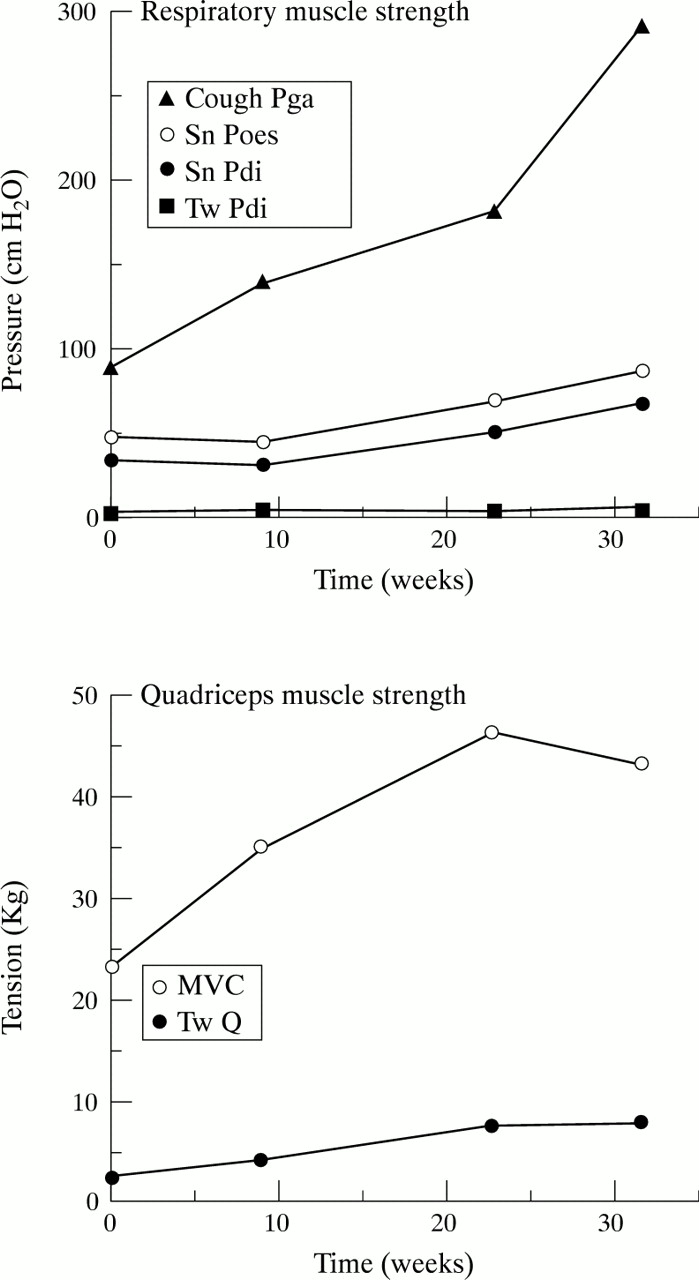
Serial strength measurements for respiratory and peripheral muscle made in a man after discharge to the ward after a prolonged intensive care unit admission after coronary artery surgery. The strength of all muscles is still increasing 4 months after discharge. Tw Pdi= transdiaphragmatic pressure elicited during bilateral magnetic phrenic nerve stimulation; Sn Pdi=transdia- phragmatic pressure during a maximal sniff; Sn Poes=oesophageal pressure during a maximal sniff; Cough Pga=gastric pressure during a maximal cough. For the quadriceps MVC=maximal voluntary contraction and Tw Q is the tension elicited by a single supramaximal magnetic stimulation of the femoral nerve.
NEUROMUSCULAR JUNCTION
Respiratory muscle weakness and, occasionally ventilatory failure, are recognised features of both myasthenia gravis and the Lambert-Eaton myasthenic syndrome. Although only a small minority of patients with myasthenia gravis come to mechanical ventilation6respiratory muscle weakness can be found in about 50% of treated patients if the appropriate investigations (for example, measurements of sniff Pdi40) are performed.41 Indeed in one study of 20 consecutive patients weakness of the diaphragm and sleep disruption were frequent findings even though the patients’ therapy was considered optimal by their neurologist.42Short term improvement in respiratory muscle strength can be secured in most such patients by the administration of intravenous edrophonium (fig 3) indicating undertreatment of the respiratory muscles. This test should therefore be considered in patients with myasthenia gravis complaining of dyspnoea even if optimal therapy with respect to the peripheral muscles has been achieved, as respiratory muscle weakness is often unrelated to peripheral muscle weakness.41Occasionally this test may precipitate a transient acute deterioration and facilities for intubation should be available. An alternative approach is to examine the effect of 3 Hz repetitive electrical phrenic nerve stimulation (and intravenous edrophonium) on the action potential of the diaphragm recorded from surface electrodes.43Similarly, potentially reversible diaphragmatic weakness is a recognised feature of the Lambert-Eaton syndrome44 which may also result in ventilatory failure and the need for mechanical ventilation.45 Identification of the syndrome may be assisted by the finding of abnormal potentiation of the action potential in response to high frequency nerve stimulation45 46 Respiratory muscle weakness also occurs in most patients with Clostridium botulinuminfection although the likelihood of requiring mechanical ventilation is dependent on the type of botulinum toxin produced; a small proportion of such patients have residual respiratory muscle weakness.47 Organophosphates are cholinesterase inhibitors which, in acute poisoning, may also result in respiratory muscle paralysis48; we are not aware of data regarding the effect of chronic organophosphate exposure on respiratory muscle function.

This patient with myasthenia gravis complained of exertional dyspnoea despite apparently optimal therapy. Transdiaphragmatic pressure was measured during a maximal sniff (upper panels) and bilateral supramaximal phrenic nerve stimulation (lower panels). Measurements were obtained before (left panels) and 10 minutes after (right panels) 2 mg intravenous edrophonium. Both indices improved and intensification of her regular medication was therefore advised; this resulted in alleviation of her symptoms.
MUSCLE
Rarely, generalised neuromuscular disease (see below) may present with acute ventilatory failure. This presentation is also well recognised for motor neuron disease.49 50 When a middle aged or older adult presents with predominant respiratory muscle weakness the differential usually lies between motor neuron disease and acid maltase deficiency.51 Separating the two may be possible using a conventional investigation for motor neuron disease (clinical examination and neurophysiology). When there is doubt, histological and, particularly, enzymatic analysis of a muscle biopsy or blood leucocytes52 is diagnostic for acid maltase deficiency.53 In younger adults inherited muscle defects— for example, nemaline myopathy, deserve consideration.51Ventilatory failure due to acute disease of muscle may be due to electrolyte disturbance, particularly hypophosphataemia or hypokalaemia, and is commonly iatrogenic. Acute respiratory muscle weakness is also a recognised feature of polymyositis,6 51 periodic paralysis, and, possibly, sarcoidosis.54
Chronic neuromuscular respiratory disease
Similar to acute neuromuscular respiratory failure, chronic respiratory neuromuscular failure may be classified according to the pathophysiological mechanism. In general, however, it is perhaps more appropriate to classify the syndrome by prognosis into those conditions with slow progression and those in which a progressive general deterioration is expected. The first group accounts for about 30% of patients receiving long term nocturnal ventilatory support in the United Kingdom55 56; within this group the most common single diagnosis is probably the postpolio syndrome. As these conditions have a good prognosis55 domicilliary mechanical ventilation is indicated and the method will be determined by the preferences of the patient and their physician and will be influenced by local expertise and resources.57 The second group presents more specific difficulties and merits more detailed consideration; this is particularly so for the two most prevalent conditions; motor neuron disease and muscular dystrophy. For both groups, however, the initial identification and assessment of chronic neuromuscular ventilatory failure is similar. Other neurological conditions also involve the respiratory muscles and may, rarely, lead to respiratory failure—for example, myotonic dystrophy,58-60 mitochondrial myopathy,61multiple sclerosis,62 limb girdle syndromes,51 and inflammatory myopathies.63
IDENTIFICATION OF CHRONIC RESPIRATORY NEUROMUSCULAR WEAKNESS
Identification of patients in chronic ventilatory failure is greatly aided by prior knowledge of the diagnosis, as is the case in muscular dystrophy; at the other extreme patients with acid maltase deficiency53 classically present with isolated respiratory muscle weakness. The diagnosis of chronic ventilatory failure is suggested by a history of fatigue, lethargy, difficulty concentrating, poor sleep and daytime somnolence (see below), and morning headache (indicating hypercapnoea). Marked dyspnoea only when lying flat is typical of isolated diaphragm paralysis, as is breathlessness on immersion in water.64 It should also be noted that, unlike patients with generalised respiratory muscle disease,65patients with isolated complete diaphragm paralysis and normal lungs are not in ventilatory failure and do not usually require ventilatory support.66 Simple tests sometimes confirm or refute the diagnosis; some—in particular the change in vital capacity on adopting the supine position2 —may be performed almost anywhere. Another test which may be helpful at the bedside is the identification of paradoxical (inward) inspiratory movement of the abdominal wall; this signifies the inability to generate about 30 cm H2O Pdi (strength reduced to at least about 30% of normal).67The approach used by our group has been described in detail elsewhere1 but is outlined below. Although the classic technique of electrical phrenic nerve stimulation68remains valuable for assessment of conduction time and amplitude, it is worth highlighting the great change brought about by the advent of magnetic phrenic nerve stimulation which, compared with electrical stimulation,69 has enabled precise and reproducible measurements of diaphragmatic strength to be obtained in various patient groups70-72 as well as selective assessment of hemidiaphragmatic function.73 The other major recent advance in the assessment of respiratory muscle strength has been the development of non-invasive tests, specifically the nasal pressure during a maximal sniff74-76 or the mouth pressure during phrenic nerve stimulation.77 78 Use of these tests in combination can obviate the need for more formal evaluation in a significant proportion of patients.79 We think that formal tests are required when respiratory muscle weakness cannot be proved or refuted using non-invasive techniques, or if precise sequential measurements are required (perhaps to assess the result of therapy). These tests involve the passage of oesophageal and gastric balloons80; the procedure is safe and practical even in elderly subjects81 and those with severe neurological disease44 72. Our basic assessment comprises measurement of dynamic compliance, sniff Pdi,40 and sniff Poes82 as well as the Pdi elicited by cervical magnetic stimulation.71 Expiratory muscle function is tested by measurement of cough gastric pressure83 and by magnetic stimulation of the thoracic nerve roots.72 84 Depending on the clinical question to be answered we would also consider separate hemidiaphragmatic assessment73 and electrophysiological evaluation using surface68 or oesophageal85electrodes as well as measurement of peripheral muscle strength.38 86 Magnetic nerve stimulation is contraindicated if the patient has a cardiac pacemaker or implanted metal (for example, after neurosurgery).
If respiratory muscle weakness of more than moderate severity is identified, then assessment of the performance of the respiratory muscle pump is warranted, at least in the form of an overnight pulse oximetry study and arterial blood gases; arterialised capillary samples drawn under local anaesthesia are an acceptable alternative to arterial blood gases.87 Polysomnography can usually be reserved for patients who deny sleep related symptoms but have proved severe weakness, or patients with sleep related symptoms in whom strength tests fail to demonstrate severe respiratory muscle weakness.
MANAGEMENT
The symptoms of respiratory failure in patients with neuromuscular disease are not usually difficult to alleviate by mechanical ventilation.57 88 For some patients nocturnal support alone will suffice, and the current approach both on the grounds of economy and practicality would be to use a positive pressure system and a nasal mask (fig 4). Bulbar disease is a contraindication for patients with stable disease, but for patients with a rapidly progressive disease in whom the primary aim is palliation this contraindication may be considered relative rather than absolute. For those with stable disease and impaired bulbar function then a tracheostomy is required. Various techniques are available for ventilatory support and practical aspects (which have been covered elsewhere89) are beyond the scope of this review. We think that, for patients with slowly progressive conditions, ventilatory support should be provided by a respiratory physician with a special interest in this condition; this will usually be in a regional centre. Conversely, for patients with rapidly progressive conditions (for example, in motor neuron disease) in whom the aim of therapy is palliative it may be considered more appropriate to institute a local treatment programme.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Patient wearing a nasal mask connected to a ventilator.
MOTOR NEURON DISEASE
As discussed earlier, respiratory failure is an occasional presentation of motor neuron disease; however, even in patients without respiratory failure indices of both inspiratory and expiratory muscle strength are often reduced at presentation59 90 and decline during the course of the disease.91 92 Moreover, the rate of decline in respiratory function is the only biological measurement which has been shown to predict survival.93Mechanical ventilation is recognised to alleviate the symptoms of ventilatory failure,94 but the nature of motor neuron disease is such that if this is achieved with the aid of a tracheostomy then the patient ultimately becomes effectively “locked in”.95 Because of ethical considerations and because in the United Kingdom there is no nationally available service which can support such patients at home, this approach does not reflect current United Kingdom practice. It is available to insured patients in some parts of the United States96 and most patients (but only half their carers) who opt for this technique are glad that they have done so.94 An alternative which we and others97-99 have used with success is the use of nocturnal nasal ventilation for symptom palliation. Our present indication for this therapy is the coexistence of symptoms and blood gas evidence of respiratory failure with evidence of respiratory muscle weakness. Relative contraindications are facial abnormalities or claustrophobia precluding the use of a mask, lack of insight into the diagnosis, and lack of a carer to set up the apparatus; we do not consider bulbar disease to be a bar to trying the therapy. Patients should be warned that this therapy may cause them to live longer in the presence of progressive disability.98 100 A further problem is that as the motor neuron disease progresses nocturnal support may be insufficient and the patient may need to use the ventilator for up to 24 hours a day.99 As with any palliative therapy nasal ventilation should be viewed as an option from a range of approaches which should be added to other locally available resources which may also be very successful.101 Low flow oxygen is commonly prescribed for such patients; whereas this may relieve some symptoms and hypoxia it usually results in a further increase in arterial CO2 102 and may therefore aggravate others.
DUCHENNE MUSCULAR DYSTROPHY
The practical problems encountered in the course of Duchenne muscular dystrophy have been previously presented103; essentially before ventilatory failure overall function can be optimised by attention to diet and prevention of contractures by physiotherapy. Not all patients with Duchenne muscular dystrophy decline at the same rate initially but the need for ventilation is heralded by a decline in the vital capacity below 1 litre, which typically occurs around the 18th birthday.104 Further data have confirmed that REM related sleep abnormalities are common in Duchenne muscular dystrophy,105 and that cough is often greatly impaired.106 It has also become accepted that nasal positive pressure support can reverse the clinical features of chronic respiratory failure.55 56 107-109 Negative pressure ventilation techniques may be initially successful but obstructive episodes are almost universal in boys treated in this way110; it should, therefore, be avoided. The use of nasal ventilation before the onset of ventilatory failure was found to confer no benefit in a multicentre French study.111 Perhaps the most difficult decision may be whether to opt for a tracheostomy; although these are widely offered in North America this is not so in the United Kingdom. As with motor neuron disease further data to clarify the desirability or otherwise of a tracheostomy are needed as are studies which might allow a more advanced prediction of when ventilatory support is likely to be required.
Sleep related disorders
Frequent disruption to sleep architecture, manifested by repetitive frequent changes in sleep stage,112 or even just autonomic disturbance,113 causes impaired daytime function. Much the most common cause of sleep disruption is obstructive sleep apnoea in which the cardinal clinical features are excessive daytime sleepiness, tiredness, difficulty concentrating, and impotence. Obstructive sleep apnoea will not be covered in detail here as authoritative sources are readily available.114-118Because of its gratifying response to treatment with nasal continuous positive airway pressure (CPAP)119 recognition of obstructive sleep apnoea is of great importance. The “gold standard” method for diagnosing obstructive sleep apnoea is formal polysomnography, but this is expensive. Thus, in our practice, formal polysomnography is not routinely offered; instead, in line with the guidelines of the British Thoracic Society, patients with the combination of suggestive symptoms and evidence of rapid nocturnal variation on domicilliary fingertip oximetry (>15 dips/hour of >4%, with a baseline >90%) are offered CPAP. This approach is highly specific for obstructive sleep apnoea but has a sensitivity of only 33%; thus symptomatic patients not meeting the desaturation criteria should proceed to polysomnography.120
In this situation polysomnography may identify obstructive sleep apnoea, which is not sufficiently severe to achieve the screening criterion; such patients may also benefit from CPAP. Alternatively the patient may have another disorder causing hypersomnolence. Periodic limb movement disorder, in which arousals follow trains of stereotyped limb movement,121 accounts for up to 5% of patients investigated for hypersomnolence.122 Clonazepam is the conventional first line therapy for periodic limb movement,123 but success has also been reported with other agents including levodopa and clonidine. Perhaps the most common remaining cause of hypersomnolence is narcolepsy,122although this diagnosis is usually suggested clinically by the other parts of the triad—cataplexy and sleep paralysis. The diagnosis of classic narcolepsy can be secured by demonstration of the HLA-DR2 antigen or its subtypes,124 but, rarely, DR2 studies are negative.125
Apnoeas arising from non-obstructive causes may be equally disruptive to sleep architecture. For patients with neuromuscular disease REM sleep is particularly hazardous because there is reduced skeletal muscle activity during REM sleep, although activity of the diaphragm is relatively preserved.126 Consequently patients who have diaphragmatic weakness experience nocturnal desaturation, which occurs predominantly and more severely in REM sleep.105 127 128Tests of inspiratory muscle strength have not been consistently found to adequately predict the occurrence of REM desaturation,105 128 but this hypothesis warrants retesting using newly available tests.1 Diminution of extradiaphragmatic respiratory muscle activity may also precipitate REM desaturation in patients with a chronically increased load on the respiratory muscle pump; this has been documented in chronic obstructive pulmonary disease129 and in restrictive lung diseases.130 REM sleep desaturations may coexist with episodes of genuine upper airway obstruction both in respiratory130 131 and neuromuscular127disease. Conventional measurements during formal polysomnography may miss rib cage and abdominal movements characteristic of upper airway obstruction. This is a particular risk in the presence of neuromuscular weakness in which the magnitude of such movements may be reduced; studies with an oesophageal balloon to measure intrathoracic pressure may occasionally be required to resolve this issue.
Respiratory consequences of neurological disease
STROKE
Acute hemiplegia due to stroke is associated with an increased risk of death due to respiratory causes; most of these deaths occur as a result of chest infections. However, pulmonary embolus has been reported in up to 9% of patients admitted with acute stroke.132 The likelihood of chest infection is increased if the patient aspirates, and intuitively an increased risk would be expected if there were hypoventilation. After stroke, diaphragmatic excursion is reduced on the hemiplegic side during voluntary but not involuntary breathing,133 134 leading to the inference that the diaphragm does not have bilateral cortical representation. Cortical magnetic stimulation studies have recently confirmed this directly.135 Similar considerations seem to apply for the parasternal intercostals,136 but to our knowledge no data exist concerning the abdominal muscles, which are the muscles chiefly responsible for cough. Small ischaemic lesions may occasionally produce isolated disorders of ventilatory control; these disorders are discussed in more detail below.
DISORDERS OF VENTILATORY CONTROL
Perhaps the most clinically prevalent neurological disturbance of respiratory control is the temporary central cessation of the drive to breathe which may follow a generalised seizure.137However, discrete anatomical lesions can, occasionally, produce characteristic disturbances of respiration. Cheyne-Stokes respiration describes a pattern in which hyperventilation periodically waxes and wanes with apnoea. It is a recognised feature of stroke but does not necessarily indicate a specific anatomical site138 and also accompanies other conditions including heart failure.139 Central neurogenic hyperventilation resulting from a pontine lesion was first systematically described in 1959 by Plum and Swanson140; in this syndrome hyperpnoea (and associated respiratory alkalosis) occurs during both wakefulness141 and sleep. For a confident diagnosis of this syndrome an anatomical or neurological basis should be demonstrated together with the absence of the more usual causes of hyperventilation (for example, pulmonary embolus or other respiratory disease). If the cause of the syndrome is treatable then recovery is possible (for example, in lymphomatoid granulomatosis142), but this is the exception; anecdotally morphine is of palliative use. A breathing pattern in which a prolonged pause follows each inspiration is described as apneustic and classically occurs because of damage to the caudal respiratory neurons in the pons; it may also occur, however, in association with lower lesions.143 Cluster breathing, in which hyperventilation rapidly alternates with apnoea, can occur in some midbrain lesions. Ataxic breathing is completely irregular both in pattern and amplitude and indicates medullary damage, as, for example in poliomyelitis140. Lesions in the lower medulla remove chemical drive without affecting voluntary control (Ondine’s curse). The classic syndrome is congenital and presents with sleep related apnoea from birth; this syndrome has a recognised association with Hirschsprung’s disease and gastro-oesophageal reflux.144Such lesions also occasionally occur in adults as a result, for example, of trauma.145 If untreated, patients with Ondine’s curse are at risk of nocturnal sudden death. Nocturnal ventilatory support is therefore recommended and for selected patients diaphragmatic pacing may be of benefit by allowing liberation from nocturnal ventilatory support as well as providing greater security against inadvertent disconnection from the ventilator. Transtentorial herniation can result in progressive abnormalities starting with Cheyne-Stokes respiration, proceeding through neurogenic hyperventilation, then eupnoea, and ultimately to an irregular gasping respiration, which is preterminal.146
BRAINSTEM AND SPINAL CORD INJURY
Patients with brainstem or high cervical cord injury are denied both voluntary and involuntary control of the respiratory muscles. These patients require ventilatory support, which is given via a tracheostomy through which tracheal suction can also be performed. For selected patients bilateral electrical pacing of the diaphragm can be an additional method of ventilatory support. In experienced hands about 50% of such patients can expect to be liberated from mechanical ventilation using this technique.147 Before pacemaker implantation cortical and cervical magnetic stimulation of the diaphragm should be performed. Diaphragmatic pacing is indicated only if the phrenic nerves are functioning but the corticodiaphragmatic pathway is interrupted. This combination is suggested by the combination of a preserved diaphragmatic EMG response to phrenic nerve stimulation but an absent or delayed response to cortical stimulation. If there is no response to peripheral stimulation then pacing will be ineffective whereas if the response to cortical stimulation is normal then eventual recovery of function is possible.148 It should be emphasised that successful application of this technique requires extremely specialised knowledge; few United Kingdom centres are currently using it.
MOVEMENT DISORDERS
Parkinson’s disease is the most common movement disorder in developed countries. Even with levodopa therapy the cause of death in such patients is commonly respiratory.149 150Characteristic findings in such patients are a reduction in both maximal static inspiratory and expiratory pressures as well as maximal inspiratory and expiratory flows.151 Moreover, such patients are unable to generate a rapid rise in peak expiratory flow which may be important for a maximally effective cough, a finding consistent with the hypokinesia typical of the condition. These abnormalities were shown to be correctable in a substantial proportion of patients by the administration of apomorphine.152Interestingly, patients with early Parkinson’s disease and normal respiratory muscle strength are markedly less able to perform repetitive bursts of respiratory muscle work than age matched controls.153 Problems in the coordination of activation of respiratory muscle also seem to be responsible for the dyspnoea which can occur in patients with dystonia.154
Upper airway flutter is sometimes found on the flow volume loops of parkinsonian patients151; this indicates upper airway dysfunction and sleep abnormalities might therefore be expected. Obstructive apnoeas are more common in parkinsonian patients, but a range of other abnormalities are also found which result in fragmented sleep architecture.155 In the related condition of multisystem atrophy, glottic narrowing during sleep has been suggested as a possible cause of sudden nocturnal death. This is not always detectable by fibreoptic examinations conducted while the patient was awake,156 157 and polysomnography may therefore be indicated in symptomatic patients. However, in another study of patients with progressive supranuclear palsy, frequent desaturations were not found.158
Neurological features in respiratory dysfunction
HYPERCAPNIA
The neurological symptoms associated with acute and chronic ventilatory failure vary, and seem to be particularly dependent on the rate of rise of CO2. In acute hypercapnia, for example, as might occur in an acute exacerbation of chronic obstructive airways disease, the most prominent symptom is drowsiness, but before this the patients may be episodically confused, and complain of headaches. Such symptoms reflect raised intracranial pressure. The physical signs include tremor, particularly of the outstretched hands; papilloedema (which in rare cases can lead to blindness159); and drowsiness advancing to coma. Seizures have also been reported. The cause of the acute encephalopathy of hypercapnia is thought in part to be due to the rapid rate of diffusion of CO2 across the blood-brain barrier. This leads to an abrupt fall in pH in the CSF, which in turn inhibits the post-synaptic receptor of the excitatory neurotransmitter glutamate.156 Overvigorous mechanical ventilation to correct hypercapnia can also cause coma and seizures, perhaps due to hyperventilation induced cerebral vasoconstriction. In chronic hypercapnia, for example, secondary to neuromuscular weakness and hypoventilation (see above), the presentation differs. Because such patients gradually retain CO2, it is rare for them to show postural tremor, asterixis, or papilloedema; arterial blood gas tensions should therefore be measured where chronic CO2retention is a possibility.
HYPOXIA
Acute hypoxia can cause transient alterations in cognitive function leading to alterations in behaviour and hallucinations. These features and others—for example, amnesia and gait disturbance—form part of the clinical picture seen with altitude sickness. Physical examination may disclose petechial retinal haemorrhages; it is also thought that petechial haemorrhages in the cerebral tissue give rise to the cerebral oedema.160 If hypoxia progresses there may be reflex depression of cardiac function, which can induce circulatory compromise that in turn induces cerebral hypoperfusion. Acute “hypoxic” brain injury follows cardiorespiratory arrest and is primarily due to cerebral ischaemia. The mechanisms underlying this type of brain damage remain an area of active study160 but it seems clear that anoxia alone does not account for the different patterns of cerebral damage seen. Specifically, the degree of associated cerebral lactic acidosis may determine whether damage is confined to the neurons or whether there is also overt infarction affecting the glial and vascular cells also.161
It has been argued that a peripheral neuropathy is associated with the chronic hypoxia associated with lung disease162 which is pathologically similar to that of diabetes mellitus.163 In one study electrophysiological evidence of neuropathy was found in 43 of 151 patients with chronic obstructive airways disease; only 30 of these patients had clinically demonstrable signs.161Additionally, another recent study164 has shown a high incidence of asymptomatic autonomic neuropathy in a group of hypoxic patients with chronic obstructive airways disease, the prognostic relevance of this is not yet clear.165 Almitrine is a respiratory stimulant which has been used in the management of patients with chronic obstructive airways disease; a mixed sensorimotor neuropathy is, however, a frequent complication of chronic administration.166
HYPERVENTILATION SYNDROME
The manifestations of the hyperventilation syndrome are protean, and misdiagnosis is not uncommon. Typically, patients complain (in rough order of frequency) of light headedness or giddiness; breathlessness; palpitations; tingling (usually but not always in the limbs), which can lead to tetany; loss of consciousness; blurring, or loss of vision; headache; nausea; unsteadiness; tremor; chest pain, or discomfort; and tinnitus.167 Other manifestations such as hallucinations and unilateral (left more common than right) somatic symptoms can lead to misdiagnoses such as epilepsy, transient ischaemic attacks, demyelination, or migraine.168 Symptom reproduction by voluntary hyperventilation has been used to diagnose hyperventilation syndrome but in a recent study in which patients were blindly subjected to both hypocapnic and isocapnic hyperventilation the presence of symptoms showed a poor relation with the presence of hypocapnia. Specifically 66% of patients who reported symptom production by hypocapnic hyperventilation also reported symptoms during isocapnic hyperventilation.169 One future possibility for the diagnosis of hyperventilation syndrome may be the relation between symptom diaries and 24 hour transcutaneous ambulatory CO2measurements.169
Several of the symptoms of hypocapnia are thought to be due to a reduction in cerebral blood flow, probably due to a change in pH rather than pCO2 and may be associated with significant cerebral hypoxia.168 The mechanisms for chest pain induced by hyperventilation are uncertain. There are many psychogenic, organic, and physiological predisposing factors that can lead to hyperventilation and often it is a combination of these factors that causes symptomatic hyperventilation.168 Of the psychogenic factors, anxiety depression phobia and panic disorder have the strongest association with hyperventilation, although the relation is complex and not necessarily causal. Many respiratory and other organic diseases—notably, mild asthma, interstitial lung disease, pulmonary embolus, and hypertension—are also associated with hyperventilation. Although physiological factors (for example, pyrexia, altitude) are rarely the sole cause of hyperventilation they can combine with other factors to induce symptoms. Progesterone can reduce PaCO2by up to 8 mm Hg in the second half of the menstrual cycle170 and this may explain the higher incidence of hyperventilation syndrome in women.
Summary
Neurological disease may result in respiratory dysfunction; however the manifestations of respiratory dysfunction in such patients may be atypical because of wider effects of their underlying condition. In the present review we have considered separately acute neuromuscular respiratory disease (as well as aspects of respiratory muscle function relevant to intensive care), chronic neuromuscular respiratory disease, sleep related disorders, respiratory consequences of specific neurological diseases, and neurological features of respiratory disease. Approaches to specific clinical problems are discussed; in many instances this can be expedited by close cooperation with a respiratory physician. We suggest that management of respiratory dysfunction in neurological disease depends critically on three factors: firstly, knowledge of when respiratory dysfunction is likely to occur; secondly, maintaining a high index of clinical suspicion (specifically apparently vague symptoms should not be uncritically attributed to the underlying neurological condition); and, thirdly, the pursuing of appropriate investigations.
References
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵
- ↵