Article Text

Statistics from Altmetric.com
The glycoprotein fibrillin is the principal component of the ciliary zonule and has an important role in the strength and elasticity of ocular connective tissues. Fibrillin polymers form the structural scaffold of extensible microfibrils1-3 which are present in ocular elastic tissues and are arranged in parallel bundles to form the zonular fibres.4 These fibrillin-rich microfibrils are morphologically identical to those which provide strength and long range elastic recoil to the connective tissues of blood vessels, lung, ligament, and dermis.5 6 In these tissues, fibrillin-rich microfibrils form a scaffold for the deposition and alignment of the elastin precursor tropoelastin during elastic fibre assembly.7 8 However, the microfibrils of the ciliary zonule as well as those of kidney glomerulus, skeletal muscle, heart, and periodontal ligament do not contain significant amounts of elastin.9
Disorders which disrupt fibrillin-rich microfibril structure or function, such as Marfan's syndrome and ectopia lentis, result in a spectrum of ocular complications. This review summarises current knowledge of fibrillin and fibrillin-rich microfibrils and their role in the eye, and discusses the pathological processes which may be involved in ocular connective tissue ageing and disease.
Fibrillin-rich microfibrils in ocular tissues
In addition to the zonules, fibrillin has been immunolocalised to the connective tissues of the anterior segment including the conjunctival, iris and ciliary body stroma, the ciliary processes, the corneal stroma and corneal epithelial basement membrane, and the endothelium of Schlemm's canal.10 In the posterior segment, fibrillin has been localised to scleral stroma, lamina cribrosa, Bruch's membrane, and choroid10; beaded microfibrils are also present in vitreous.11-13 The precise role of fibrillin-rich microfibrils in all these ocular tissues is not defined, but they may regulate development and confer strength and elasticity to connective tissues. Fibrillin present in the equatorial region of the lens capsule allows anchorage of the zonular fibres.14
Composition and ultrastructure of fibrillin-rich microfibrils
Fibrillin exists as two isoforms, fibrillin-1 and fibrillin-2 (Fig1); the gene for human fibrillin-1 (FBN-1) has been localised to chromosome 15q15–21 and the gene for human fibrillin-2 (FBN-2) to chromosome 5q23–31.15 Fibrillin-1 and fibrillin-2 have distinct but overlapping spatiotemporal tissue distributions, and it is unclear if they form separate populations of microfibrils or if they can coexist in the same microfibril.16-18 It has been suggested that fibrillin-1 may provide force bearing structural support to tissues, whereas fibrillin-2 may play an important part in the initiation of elastogenesis.18 The microfibrils of the ciliary zonules (Figs 2 and 3) are almost exclusively composed of fibrillin-1.4 9

Schematic diagram of multidomain structure of fibrillin-1 and fibrillin-2.
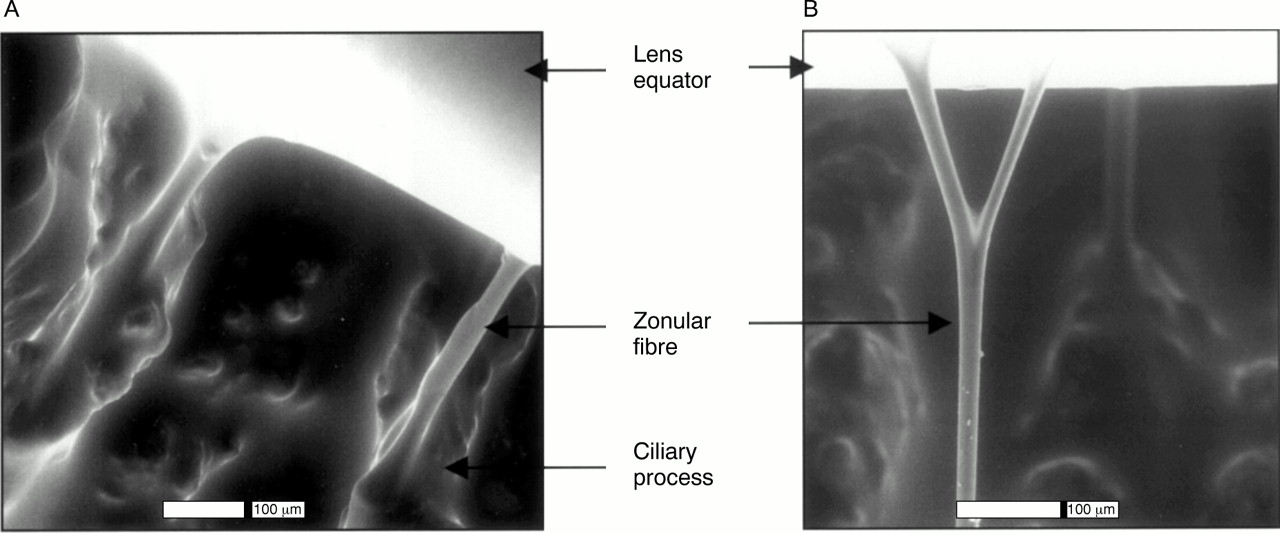
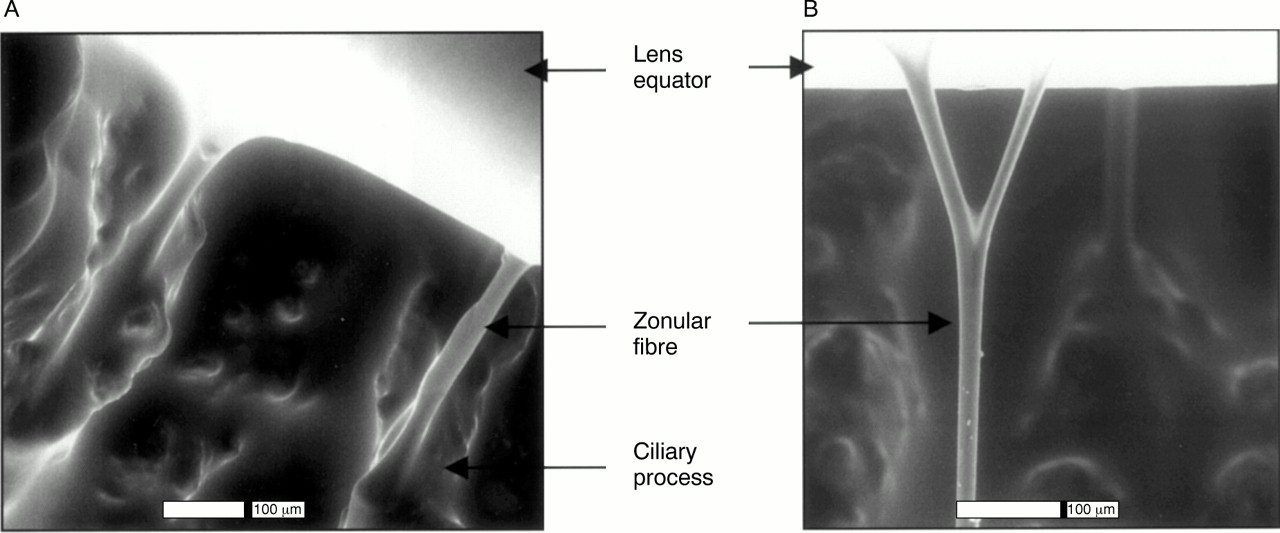
Environmental scanning electron microscopy of normal hydrated human zonules. (A) Zonular fibres arising from ciliary processes. (B) Branching of a zonular fibre.

Transmission electron microscopy (TEM) of human zonular microfibrils, and effects of matrix metalloproteinase treatment. Normal human zonular specimens from a 54 year old were incubated for 3 hours at 37°C in the presence of 10 mM calcium chloride and hyaluronidase to remove adherent vitreous, with and without MMP-13, before fixation and TEM with uranyl acetate and lead citrate stain. (A) and (B) are untreated zonules which consist of dense and regular, parallel striated microfibrils. (C) and (D) are MMP-13 treated zonules. The zonular microfibrils are fragmented and irregularly and loosely arranged. Magnification: (A) and (C) ×20 000; (B) and (D) ×68 000.
Both fibrillin-1 and fibrillin-2 have a molecular structure of multiple protein subunits,1 the majority of which are epidermal growth factor (EGF)-like domains (Fig 1). Most of these EGF-like domains are capable of binding calcium which maintains the fibrillin molecule in an extended, rod-like conformation19 20 and stabilises it against degradation by proteases.21Interspersed with the EGF-like domains are protein subunits containing eight cysteine residues (Fig 1). One of the most striking differences between fibrillin-1 and fibrillin-2 is in the amino acid composition of the amino terminal region which may serve as a molecular hinge22 and may also mediate fibrillin dimer formation during microfibril assembly.23 24 The extreme carboxy terminal region of the fibrillin molecule is cleaved off before assembly into microfibrils.25-27
Isolated fibrillin-rich microfibrils have a diameter of 10–12 nm and a characteristic “beads on a string” appearance when imaged by rotary shadowing or scanning transmission electron microscopy (STEM)2 28 (Fig 4). The average untensioned interbead distance is 56 nm but this increases when the microfibrils are subject to tension.31 The arrangement of fibrillin molecules within microfibrils is unknown. Recent evidence suggests that fibrillin molecules may form dimers before assembly into microfibrils,23 24 and an association between the overlapping carboxy and amino termini of adjacent fibrillin molecules allows linear growth of the microfibril, stabilised by intramolecular crosslinks.32 33 However, several other models of microfibril assembly are possible, including a parallel, unstaggered arrangement of fibrillin molecules within the microfibrils.34 Compaction of fibrillin molecules within microfibrils may allow microfibril extensibility, so that molecules can “unravel” in the interbead region.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Scanning transmission electron microscopy of an isolated human zonular microfibril. Bar = 100 nm. Zonular microfibrils were biochemically isolated using an established method29 30 by incubation with collagenase and hyaluronidase in the presence of protease inhibitors, followed by size fractionation. The excluded volume (Vo) contained fibrillin-rich microfibrils.
An increasing number of other proteins have been identified in association with fibrillin containing microfibrils in addition to fibrillin-1 and fibrillin-2. In most instances, it is unclear whether the protein is an integral structural component of the microfibril or is just adherent to its surface. The relative proportions of most of these proteins varies between tissues, and they may thus contribute to the tissue specific structural and functional characteristics of fibrillin-rich microfibrils. Microfibril associated glycoprotein-1 (MAGP-1), also designated microfibril associated protein-2 (gene symbol MFAP-2),35 has been shown by immunoelectron microscopic techniques to be associated with the bead regions of zonular and vitreous microfibrils.36 Microfibril associated protein-1 (MFAP-1) has been immunolocalised to microfibrils of chick aorta, bovine nuchal ligament, and human ocular zonules.37 Emilin (elastin microfibril interface located protein) is a glycoprotein which is abundant at the elastin-microfibril interface and has been immunolocalised in the eye to the zonules, to elastin-free microfibrils of the cornea and to Descemet's membrane.38 Microfibril associated molecules may mediate cell adhesion to microfibrils and may stabilise the interactions of microfibrils with other structural elements of the extracellular matrix.
Ocular conditions associated with abnormalities in fibrillin-rich microfibrils
Microfibril abnormalities occurring as a result of mutations in the gene for fibrillin-1 are manifest as a spectrum of disease phenotypes ranging from severe, lethal neonatal Marfan's syndrome to “simple” ectopia lentis. There is no clear correlation between genotype and phenotype, with the exception of a clustering of fibrillin-1 mutations associated with severe neonatal Marfan's syndrome.39 Approximately 60% of patients with Marfan's syndrome have ectopia lentis.40 Familial ectopia lentis also occurs in patients in whom the clinical criteria for Marfan's syndrome are not fulfilled, although some of these patients have systemic features such as arachnodactyly or tall stature.41-43
The structural consequences of microfibrillar abnormalities have been demonstrated by examination of ocular tissues from patients with ectopia lentis and Marfan's syndrome. Ectopia lentis zonular fibres have been shown to be reduced in number,44 thin, stretched, and irregular in diameter,14 45 and inelastic and easily broken when compared with normal controls.45 46 The insertion of zonular fibres onto the lens capsule has also been noted to be abnormal in patients with Marfan's syndrome and ectopia lentis, with reduction in the amount of fibrillin present as demonstrated by immunostaining.14Ultrastructural abnormalities of fibrillin-rich microfibrils of ciliary zonules have also been demonstrated in ectopia lentis and Marfan's syndrome; the microfibrils are loosely arranged and disorganised within the zonular fibres, with fragmentation and a variable interbead periodicity.46 There are several possible mechanisms by which the observed zonular abnormalities in Marfan's syndrome and ectopia lentis may result from mutations in fibrillin. Structurally weak zonules may result from a reduction in the amount of fibrillin-1 synthesised and secreted during fetal development or from incorporation of mutant fibrillin-1 into microfibrils. However, these mechanisms do not explain the intrafamilial phenotypic variability between patients with the same fibrillin-1 mutation or the progressive nature of some of the clinical manifestations. Recently, it has been suggested that proteolytic degradation of microfibrils may contribute to the observed structural and functional changes in connective tissues and zonules30 47 (see below).
Marfan's syndrome and familial ectopia lentis are associated with a spectrum of other ocular abnormalities including axial myopia, presenile cataract, increased prevalence of open angle glaucoma,48 strabismus,49 corneal flattening and hypoplasia of the ciliary muscle and iris, resulting in miosis,50 51 and elongation of the ciliary processes45 52 and pars plana (DMcL, personal observation).
Ectopia lentis is seen in association with a variety of other conditions which must be differentiated from “simple” ectopia lentis or Marfan's syndrome and which affect cysteine metabolism or development of the eye, or cause mechanical zonular disruption. One such condition is the Weill-Marchesani syndrome, a congenital connective tissue disorder characterised by short stature, brachydactyly, microspherophakia, glaucoma, and ectopia lentis which may be inherited in an autosomal recessive or dominant manner; the autosomal dominant form has been linked to the gene for fibrillin-1 on chromosome 15q21.1 and immunohistochemical staining of skin sections from an affected family shows a decrease in fibrillin staining compared with normal controls.53 Another is the pseudoexfoliation syndrome which is also occasionally associated with lens dislocation; the pseudoexfoliative material has been shown to have strong immunoreactivity for fibrillin,54-56 suggesting that this condition may also result from an abnormality of fibrillin-rich microfibrils. It can be speculated that abnormalities of zonular proteolytic degradation may result in zonular weakness and release of microfibril constituents seen as pseudoexfoliative material in the anterior chamber.
Proteolytic degradation of fibrillin-rich microfibrils in ocular ageing and disease
The extent of physiological zonular degradation during development and ageing is unknown although the zonular insertion onto the lens capsule has been observed to shift progressively anteriorly with age,57 suggesting that the ageing zonules undergo remodelling. Other factors may contribute to this shift, however, such as the changing curvature of the lens and contraction of the anterior capsule.58 Proteolytic damage to zonular microfibrils potentially contributes to these observed ageing changes as well as to the pathogenesis of zonular dysfunction in patients with Marfan's syndrome and ectopia lentis.
The zonules have been known for many years to be susceptible to degradation by serine proteases59 as demonstrated by the use of chymotrypsin for zonulysis during intracapsular cataract surgery. The serine proteases are a large group of enzymes which are secreted by inflammatory cells and whose activity is regulated by inhibitors such as α1 antitrypsin in the plasma and tissue thrombospondin. The zonules are normally exposed to low levels of serine proteases in the aqueous and, potentially, to proteases released by inflammatory cells.60 61 Both fibrillin-1 molecules and fibrillin-rich microfibrils are susceptible to degradation by serine proteinases such as neutrophil elastase, chymotrypsin, and trypsin.21 62 Calcium binding stabilises fibrillin against such proteolytic degradation.21 Mutations in fibrillin-1 that affect calcium binding may therefore increase fibrillin-1 susceptibility to proteolytic degradation by reducing calcium binding affinity and inducing conformational changes which expose cryptic protease cleavage sites.
Matrix metalloproteinases are another important class of proteases which are widely expressed in ocular tissues and which may be involved in ocular development, physiological remodelling, and wound healing. They are secreted by a wide range of cell types including mesenchymal cells such as fibroblasts and inflammatory cells such as macrophages and neutrophils. Several matrix metalloproteinases are implicated in the turnover of elastic connective tissues owing to their elastinolytic activity,63 but the contribution of matrix metalloproteinases to turnover and degradation of fibrillin-rich microfibrils in zonules and other connective tissues has only recently been reported. Matrix metalloproteinases are implicated in matrix remodelling associated with normal mammalian development and growth, and are also thought to participate in the accelerated breakdown of extracellular matrix in diseases such as arthritis, atherosclerosis, tissue ulceration, and tumour invasion and metastasis. The proteolytic capacity of matrix metalloproteinases is normally tightly controlled by regulation of the rate of mRNA transcription, by activation of the pro-enzyme and also by naturally occurring inhibitors such as tissue inhibitors of metalloproteinases (TIMPs) and the plasma protein inhibitor α2 macroglobulin.63 An imbalance in ocular matrix metalloproteinase activity may underlie dysregulation of angiogenesis, abnormalities of matrix turnover, tumour invasion, and abnormalities of wound healing in the eye.
Matrix metalloproteinases and their inhibitors are present in normal aqueous,61 64 where they may play a part in remodelling of the tissues bordering the anterior chamber. It can be speculated that aqueous matrix metalloproteinase activity will be increased in intraocular inflammation owing to release from inflammatory cells. Regulation of matrix metalloproteinase mediated extracellular matrix turnover in the trabecular meshwork is thought to be important for maintenance of normal aqueous humour outflow, and an imbalance in matrix metalloproteinase activity may be implicated in the pathogenesis of open angle and neovascular glaucomas.65 66
The widespread distribution of matrix metalloproteinases in the eye and their potential involvement in many abnormal ocular conditions suggest that zonular microfibrils may be exposed to matrix metalloproteinases under physiological and pathological conditions. Matrix metalloproteinases are present in the cornea (where they are produced by the corneal epithelium, stromal keratocytes, and fibroblasts67 68) and are thought to be important in corneal ulceration and wound healing.69 70 Matrix metalloproteinases and TIMPs may be involved in postoperative conjunctival wound healing.71 Matrix metalloproteinase-2 (also called gelatinase A) is upregulated in the sclera in an experimental model of myopia, and may be involved in scleral remodelling associated with axial elongation of the eye.72Matrix metalloproteinases and TIMPs are present in vitreous73 74 and are potentially involved in vitreous liquefaction which occurs in ageing and pathological states.75 Matrix metalloproteinase activity may have a role in retinal neovascularisation in proliferative diabetic retinopathy76 77 and the development of proliferative vitreoretinopathy after surgery for retinal detachment.78Matrix metalloproteinases and TIMPs are expressed by retinal pigment epithelial cells,74 79 which may be the source of the increased TIMP levels found at the macula in age related macular degeneration.80 A mutation in TIMP-3 is associated with Sorsby's fundus dystrophy, an autosomal dominant condition in which lipid-rich deposits accumulate in Bruch's membrane.81 The choroidal neovascularisation and subretinal haemorrhages seen in this condition are consistent with a imbalance in the regulation of angiogenesis by TIMPs.82
Recent work has demonstrated that members of the matrix metalloproteinase family of enzymes can degrade recombinant fibrillin-1 molecules and disrupt fibrillin-rich microfibrils,30 as well as disrupting the ultrastructure of intact zonular bundles (Fig3). Matrix metalloproteinases may therefore be important in physiological and pathological turnover of fibrillin microfibrils in the ocular extracellular matrix and the ciliary zonule. The aortae of patients with Marfan's syndrome demonstrate strong immunoreactivity for elastolytic matrix metalloproteinases adjacent to areas of cystic medial necrosis,83 suggesting that matrix metalloproteinases may have a role in the pathogenesis of aortic aneurysms in Marfan's syndrome. In addition, a mutation identified in a family with familial ectopia lentis has been shown to alter the proteolytic degradation patterns of fibrillin-1 molecules by several matrix metalloproteinases, probably by exposure of a cryptic cleavage site.30 This mutation (E2447K)41 43 occurs within a calcium binding EGF-like domain of fibrillin-1 and is predicted to reduce fibrillin-1 calcium binding and alter molecular conformation. The proteolytic susceptibility of zonular microfibrils containing mutant allele products would therefore be expected to be increased, resulting in progressive zonular damage and eventual ectopia lentis. However, other ectopia lentis causing mutations in fibrillin-1 do not alter degradation patterns (JLA, unpublished data) so other mechanisms, such as proteolytic degradation of reduced amounts of microfibrils or increased overall activity of matrix metalloproteinases, must also lead to functional insufficiency of zonules. The presence of matrix metalloproteinases in normal aqueous suggests that they may cause progressive zonular degradation throughout life, and are a potential cause of weakened zonules with age as seen during cataract surgery.
Future research directions and therapeutic implications
Future research will determine whether matrix metalloproteinase levels are upregulated in the aqueous of patients with Marfan's syndrome and ectopia lentis and whether there are individual variations in matrix metalloproteinase activity which might explain the intrafamilial variability in the phenotypic manifestations of fibrillin-1 mutations. It can be predicted from the position of matrix metalloproteinase cleavage sites identified within the fibrillin-1 molecule84 that microfibril degradation would result in stable amino and carboxy terminal fibrillin-1 fragments. These fragments may affect cellular behaviour and could potentially be involved in positive feedback leading to upregulation of matrix metalloproteinases. Although stable intermolecular crosslinks may prevent release of fibrillin fragments from damaged microfibrils,32 their presence in blood or in aqueous could potentially be used as a marker of progressive microfibril damage in Marfan's syndrome.
Altered extensibility of zonular fibrillin-rich microfibrils as a result of proteolytic damage potentially contributes to presbyopia. Variations in aqueous matrix metalloproteinase levels may develop with age or between individuals and may correlate with zonular instability, thus explaining why variations in zonular strength occur between otherwise normal patients. Further work on the pseudoexfoliation syndrome will determine if altered turnover of fibrillin-rich microfibrils is implicated in the pathogenesis of this disorder, with important implications for therapeutic intervention.
Potential therapeutic strategies for prevention of ectopia lentis and other ocular manifestations of proteolytic damage include the use of synthetic matrix metalloproteinase inhibitors. The eye would provide an ideal opportunity to localise the application of matrix metalloproteinase inhibitors in order to prevent or to slow the progression of ectopia lentis in susceptible patients. Alternatively, genetic manipulation of TIMP or matrix metalloproteinase levels may underlie a range of therapeutic strategies in the future.
Acknowledgments
JLA was supported by a Wellcome Trust Vision research training fellowship and CMK is supported by the Medical Research Council. We would like to thank Mike Sherratt, Chris Gilpin, and Carolyn Jones for the electron micrographs.
References
Contributors please note:
Communications fromall countries except the UK and Republic of Ireland should be sent to Professor C Hoyt, Editor, British Journal of Ophthalmology, University of California, Department of Ophthalmology, 10 Kirkham Street, K 301, San Francisco, CA 94143-0730, USA (tel: 001 415 502-6871; fax: 001 415 514-1521).
Manuscripts from theUK and the Republic of Ireland should be sent to Professor Andrew Dick, UK Editor, British Journal of Ophthalmology, Division of Ophthalmology, University of Bristol, Lower Maudlin Street, Bristol BS1 2LX (tel: +44 (0) 0117 929-4496; fax: +44 (0)117 929-4607).