Article Text

Abstract
Takayasu arteritis is a well known yet rare form of large vessel vasculitis. This review details the history, clinical features, differential diagnoses, classification, and immunology of the disorder. Suppression of inflammation and preservation of vascular competence are the aims of treatment. As with any rare disease, randomised controlled treatment trials are either lacking or based on small patient numbers, making management decisions difficult. Current evidence based treatments are presented and discussed.
- arteritis
- pathogensis
- Takayasu
- treatment
- ACR, American College of Rheumatology
- CRP, C reactive protein
- ESR, erythrocyte sedimentation rate
- HLA, human leucocyte antigen
- IL, interleukin
- MRA, magnetic resonance angiography
Statistics from Altmetric.com
- ACR, American College of Rheumatology
- CRP, C reactive protein
- ESR, erythrocyte sedimentation rate
- HLA, human leucocyte antigen
- IL, interleukin
- MRA, magnetic resonance angiography
Takayasu arteritis, also known as pulseless disease, occlusive thromboaortopathy, and Martorell syndrome,1 is a chronic inflammatory arteritis affecting large vessels, predominantly the aorta and its main branches. Vessel inflammation leads to wall thickening, fibrosis, stenosis, and thrombus formation. Symptoms reflect end organ ischaemia. More acute inflammation can destroy the arterial media and lead to aneurysm formation.2 Early reports suggested that the disease was confined to females from Eastern Asia, but it has now been recognised worldwide in both sexes, although disease manifestations vary between populations. The female to male ratio appears to decline from Eastern Asia towards the West.
HISTORY
Published descriptions of this arteritis date back as far as 1830.2 Yamamoto described the case of a 45 year old man with persistent fever who developed impalpable upper limb and carotid pulses associated with weight loss and dyspnoea.3 In 1905 Takayasu, professor of ophthalmology at Kanazawa University Japan, presented the case of a 21 year old woman with characteristic fundal arteriovenous anastamoses.4 In the same year, Onishi and Kagosha each described similar cases associated with absent radial pulses.3 In 1920, the first postmortem case of a 25 year old woman demonstrated panarteritis and suggested that the fundal appearances resulted from retinal ischaemia.2 In 1951, Shimizu and Sano summarised the clinical features of this “pulseless disease”.5
INCIDENCE
Takayasu arteritis is rare, but most commonly seen in Japan, South East Asia, India, and Mexico. In 1990, it was included in the list of intractable diseases maintained by the Japanese government,2 and to date 5000 patients have been registered. A study of North American patients by Hall et al found the incidence to be 2.6/million/year.6 The UK incidence is unknown.
CLINICAL FEATURES
The clinical features have been well documented by cohort studies of over 570 patients from different countries.1,6–13 Manifestations range from asymptomatic disease found as a result of impalpable pulses or bruits, to catastrophic neurological impairment. A two stage process has been suggested with a “pre-pulseless” phase characterised by non-specific inflammatory features, followed by a chronic phase with the development of vascular insufficiency, in some cases accompanied by intermittent flares, although not all patients conform to this pattern.6
“As the inflammation progresses and stenoses develop, the more characteristic features become apparent, influenced by the development of collateral circulation”
The disease commonly presents in the 2nd or 3rd decade of life, often with a delay in diagnosis from the onset of first symptoms of months to years. In one of the largest cohorts (n = 107) 80% of patients were between 11 and 30 years, 77% had disease onset between the ages of 10 and 20 years, with time from onset of symptoms to diagnosis of two to 11 years in 78%.1 A study of 88 patients from India9 gave a mean (SD) age at symptom onset of 24.0 (8.8) years and mean (SD) age at diagnosis of 28.3 (9.9) years. The National Institute of Health study by Kerr et al suggested that the delay in diagnosis was longer in juveniles, being up to four times that of adult patients.10 However, data from India12 looking at patients aged under 18 years demonstrated a delay of only 2.5 to 5.5 months. This discrepancy presumably relates to the difference in disease incidence between the two populations, which results in differences in awareness. The clinical features and progress of young patients with Takayasu arteritis appear to be very similar to those of adults.12
Non-specific features include fever, night sweats, malaise, weight loss, arthralgia, myalgia, and mild anaemia.6 As the inflammation progresses and stenoses develop, the more characteristic features become apparent, influenced by the development of collateral circulation. Stenotic lesions predominate9,10 and tend to be bilateral. Nearly all patients with aneurysms also have stenoses and most have extensive vascular lesions.
CHARACTERISTIC FEATURES
-
Diminished or absent pulses in 84–96% of patients1,9 associated with limb claudication and blood pressure discrepancies.
-
Vascular bruits in 80–94% of patients,1,6,10 often multiple, and particularly affecting the carotids, subclavian, and abdominal vessels.
-
Hypertension in 33–83% of patients,1,6,7,10,12 generally reflecting renal artery stenosis, which is seen in 28–75% of patients.1,10,12
-
Aortic regurgitation resulting from dilatation of the ascending aorta, separation of the valve leaflets, and valve thickening in 20–24%.9,10
-
Congestive cardiac failure associated with hypertension, aortic regurgitation, and dilated cardiomyopathy.9
-
Neurological features secondary to hypertension and/or ischaemia, including postural dizziness, seizures, and amaurosis.
-
Pulmonary artery involvement in 14–100% of patients, depending on the method used to assess pulmonary vasculature. Oligaemic lung fields on plain chest x ray correlate with pulmonary vasculopathy in approximately a third of cases.14 Pulmonary artery disease shows little correlation with the systemic pattern of arterial involvement,7,14 but can be useful in the differential diagnosis by helping to confirm Takayasu arteritis.
-
Other symptoms include dyspnoea, headaches, carotodynia, myocardial ischaemia, chest wall pain, and erythema nodosum.
Variable disease presentation between different populations is well illustrated by Moriwaki et al in their study of Indian and Japanese patients.11 The Japanese patients (n = 80) were predominantly female (96%), presenting with dizziness, vertigo, pulselessness, more prolonged and severe inflammation, and more aortic regurgitation, reflecting involvement of the aortic arch and its main branches. This contrasted with the Indian patients (n = 102), 37% of whom were male. They tended to present with headache, hypertension, and left ventricular hypertrophy as a result of vasculitis affecting the abdominal aorta and renal vessels. However, most patients in both countries had diffuse disease.
DIAGNOSIS AND DIFFERENTIAL DIAGNOSIS
From the more typical features of Takayasu's arteritis, the American College of Rheumatology (ACR) defined specific diagnostic criteria for this disorder in 1990 (table 1).15 Angiography remains the gold standard for diagnosis (figs 1, 2). Assessment of pulmonary vasculature by angiography is not universally recommended, being reserved for patients with symptoms of pulmonary hypertension.10 Doppler ultrasound is a useful non-invasive procedure for the assessment of vessel wall inflammation. In view of the vessels involved, histological diagnosis is usually impractical and histological assessment is limited to those cases undergoing revascularisation procedures.
1990 ACR criteria for the classification of Takayasu arteritis15
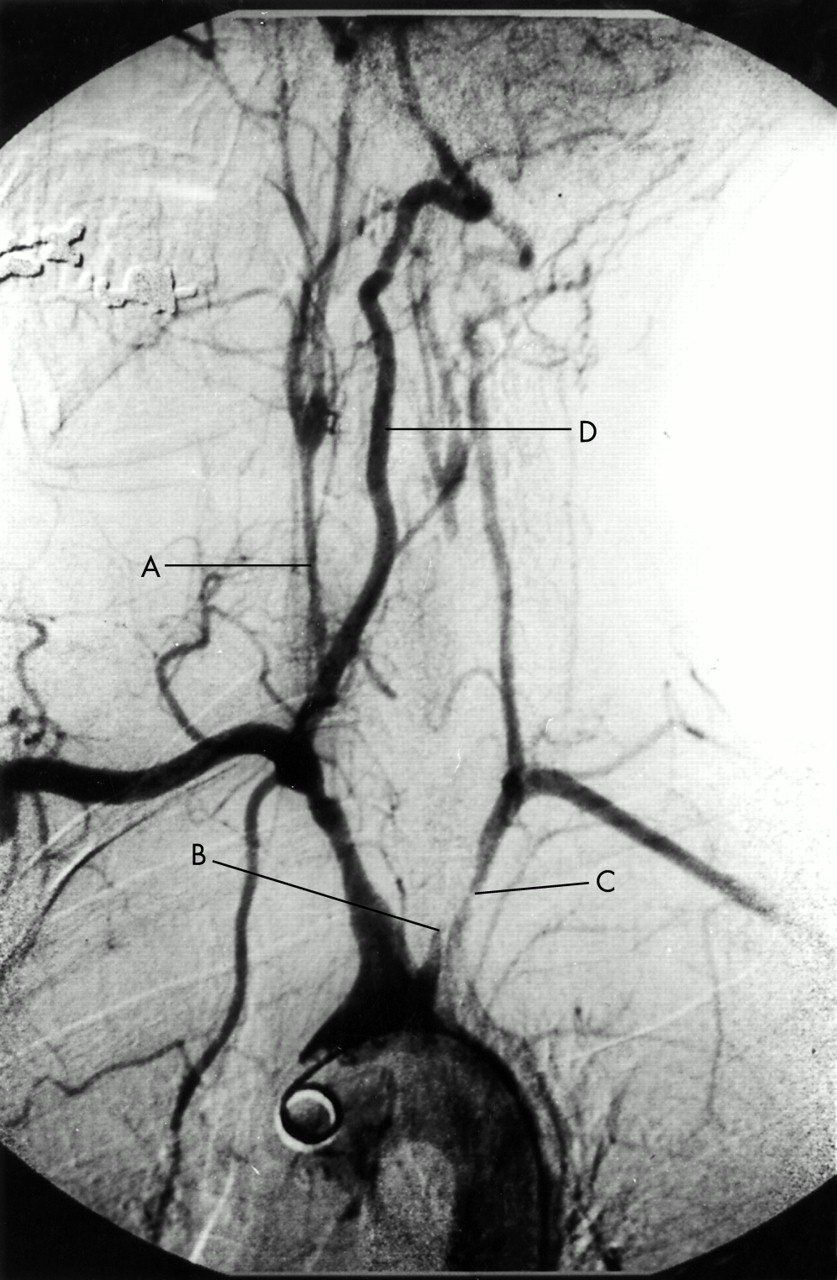
Arch aortogram demonstrating (A) a severely narrowed right common carotid artery, (B) occlusion of the left common carotid artery and, (C) proximal stenosis of the left subclavian artery. (D) The right vertebral artery provides the dominant cerebral supply.

{kind=link}
{kind=link}
Arch aortogram demonstrating severe involvement of all extracranial vessels; the descending thoracic aorta appears to be normal.
The differential diagnoses include other causes of large vessel vasculitis: inflammatory aortitis (syphilis, tuberculosis, lupus, rheumatoid arthritis, spondyloarthropathies, Behçet's disease, Kawasaki disease, and giant cell arteritis); developmental abnormalities (coarctation of the aorta and Marfan syndrome), and other aortic pathologies, such as ergotism and neurofibromatosis. Most of these have specific features that enable diagnosis, but tuberculosis has remained an important differential and possible aetiological factor. However, tuberculous aortitis tends to cause erosion of the vessel wall with the formation of true or false aneurysms, particularly affecting the descending thoracic and abdominal aorta. Dissection and rupture are important complications rather than the stenoses typical of Takayasu arteritis. The incidence of rupture and bleeding complications of aneurysmal Takayasu arteritis is low. Syphilis tends to affect an older age group, with calcification, sparing the descending thoracic aorta, and stenoses are not a feature.9 Hypertension as a result of fibromuscular dysplasia is an important differential diagnosis.
Although similar in many respects, including aortic involvement in 10–15% of patients with giant cell arteritis, Michel et al suggest that giant cell arteritis and Takayasu arteritis can be differentiated on clinical grounds. In a study of 280 patients, 217 with giant cell arteritis and 63 with Takayasu arteritis identified through the ACR vasculitis criteria databank, they found that age of 40 years at disease onset was the single most discriminatory factor. Excluding age from the analysis, ethnic background and clinical signs of upper limb vascular insufficiency, shoulder stiffness, and scalp tenderness were variables that led to correct diagnoses in 95% of patients.16
CLASSIFICATION
An attempt has been made to classify the disease on the basis of angiographic findings. The early system, revised by Lupi-Herrera et al in 1977,1 has been superseded by the new classification of Takayasu arteritis (table 2).11 These systems are useful in that they allow a comparison of patient characteristics according to the vessels involved and are helpful in planning surgery, but they offer little by way of prognosis.
New angiographic classification of Takayasu arteritis, Takayasu conference 199411
Most patients in the large series studied have diffuse disease.
The natural history of any disorder can only be elucidated by following patients in the absence of specific treatment. Ishikawa defined clinical groups based on the natural history and complications of the disease.7 The four most important complications were defined as Takayasu retinopathy, secondary hypertension, aortic regurgitation, and aneurysm formation, each being graded as mild/moderate or severe at the time of diagnosis. Four grades of disease are described (table 3).
Ishikawa clinical classification of Takayasu arteritis7
“Tuberculosis has remained an important differential and possible aetiological factor”
Ishikawa retrospectively studied 54 Japanese patients over six months to 18 years of follow up between 1957 and 1975. The overall five year survival rate after diagnosis was 83.1%. Seven patients died within five years of diagnosis, all were in groups IIB and III, and deaths were mostly from cerebrovascular disease and congestive cardiac failure. All patients with aortic regurgitation were in group III. The five year survival rate in combined groups IIB and III was 70%, compared with 100% in group I. Five acute events occurred in the survivors during follow up, three of five occurring in patients from groups IIB and III. No acute event occurred in patients from group I. Nineteen of the 54 patients were treated with steroids.
The experience from India supports this classification for prognostic assessment.9 Cumulative survival at five years after disease onset was 91%, after 10 years the figure was 84%, whereas event free survival figures were 74.9% and 64%, respectively. Patients with a single mild complication or no complication at diagnosis had a five year event free survival of 97%, compared with 59.7% in patients with a single severe or multiple complications. No deaths occurred in patients in groups I and IIA, whereas 19.6% of patients in groups IIB and III died during follow up, mostly from cerebrovascular disease and cardiac failure. Twenty two major non-fatal events occurred during follow up, with 20 of 22 occurring in groups IIB and III. In this study, 63 of 88 patients received no specific disease modifying treatment. Other studies, which have included patients treated more aggressively, give five year survival rates of 90–94%.6,13 Therefore, classification according to this system appears to give useful prognostic information at diagnosis and may help to guide treatment.
HISTOLOGY, IMMUNOLOGY, AND PATHOGENESIS
Macroscopically, in the chronic phase, the aorta is thickened secondary to fibrosis of all three vessel layers. The lumen is narrowed in a patchy distribution, often affecting multiple areas. If disease progression is rapid, fibrosis can be inadequate with subsequent aneurysm formation. The intima may be ridged, with a “tree bark” appearance, a feature common to many aortitides.17
Microscopically, the vasculitis may be divided into an acute florid inflammatory phase and a healed fibrotic phase. In the acute phase a vasa vasoritis is seen in the adventitia. The media is infiltrated by lymphocytes and occasional giant cells with neovascularisation. Mucopolysaccharides, smooth muscle cells, and fibroblasts thicken the intima. In the chronic phase there is fibrosis with destruction of elastic tissue. Similar histopathological findings are also seen in giant cell arteritis; therefore, biopsy results may not differentiate between these two vasculitides. Clinical features usually allow correct diagnosis,16 but difficulties can be envisaged in older patients with Takayasu arteritis when the timing of disease onset is uncertain.
Recent investigation of the cellular composition of the aortic wall18 has shown neovessels in the deep intima associated with the adventitial vasa vasorum. T cells and dendritic cells, with few B cells, granulocytes, and macrophages, surrounded the vessels. The media contained acellular fibrous tissue, with bundles of neovascularisation and sparse smooth muscle cells. Inflammation was most prominent in the adventitia, with infiltration of B and T cells. In half of the cases these formed nodules, with central B cells and peripheral T cells in close proximity to antigen presenting dendritic cells. Granulocytes were located outside of the nodules and granulocyte destruction was observed. No giant cells were seen.
Infection has been considered to play a role in the pathogenesis of Takayasu arteritis. Tuberculosis has been particularly implicated in view of the high prevalence of infection, past or present, in affected patients,1,9 largely from endemic areas. More recently, viral infection is being investigated as a trigger of vasculitis.19
Seko et al have reported that γδT cells, αβT cells (CD4 and CD8), and natural killer cells play an important role in the vascular injury.20 The 65 kDa heat shock protein to which γδT cells respond is strongly expressed in the aortic tissue of patients with Takayasu arteritis. They have previously found restricted VαVβ gene usage of the αβT cell receptor, suggesting that a specific antigen was being targeted. More recently, they have reported restricted usage of the VγVδ genes in the infiltrating γδT cells, supporting their hypothesis, along with the expression of various costimulatory molecules necessary for T cell activation.
Takayasu arteritis has been associated with different human leucocyte antigen (HLA) alleles in different populations.21–23 For example, in Japan and Korea there is a clear association with the extended haplotype: HLA B*52, DRB1*1502, DRB5*0102, DQA1*0103, DQB1*0601, DPA1*02-DPB1*0901.21 Sequence analysis has shown that some of the alleles share specific epitopes and it may be that the epitopes are more important as a disease susceptibility factor than the allele in which they are found. The HLA association is thought by some to strengthen the argument in favour of an autoimmune pathogenesis. However, no specific autoantigens have yet been identified and for any adaptive immune response to occur, whether against exogenous or endogenous antigen, presentation of antigen to T cells in the context of the major histocompatibility complex is central.
“A study reported in 1998 concluded that no known serological test was able to supplant vascular histopathology in determining disease activity”
Several studies have examined the acute phase response in Takayasu arteritis. Ishikawa found that the erythrocyte sedimentation rate (ESR) was raised in 29 of 54 patients studied,7 with an equal distribution in the four disease categories. Higher values were seen in the younger patients, declining with age, perhaps representing the natural history of the disease. Hall et al found that the ESR was raised in three quarters of 32 cases, and that it showed an excellent correlation with treatment effect.6 However, Kerr et al concluded that the ESR was not a consistently reliable marker of disease course, being raised in 72% with active disease but also in nearly half of patients in clinical remission.10 In their study, 44% of arterial biopsy specimens obtained from patients with clinically inactive disease demonstrated vasculitis, suggesting that disease activity may be underestimated, a view also supported by P Bacon (personal communication, 2001).
This inconsistency has led to a search for better serological markers. A study reported in 199824 concluded that no known serological test was able to supplant vascular histopathology in determining disease activity. This study compared 29 patients (22 with clinically inactive disease and seven with clinically active vasculitis) with 26 healthy control volunteers; no serological test reliably distinguished healthy volunteers from patients with active disease. The markers assessed included ESR, C reactive protein (CRP), tissue factor, von Willebrand factor, thrombomodulin, and tissue plasminogen activator, in addition to various adhesion molecules. The numbers with clinically active disease were small and again may have been underestimated in the absence of histological assessment. ESR and CRP values were not directly compared. Although disease activity may not be discriminated by these markers at a single point in time, for individual patients the use of a given parameter longitudinally may still be of value.
Serum concentrations of the pro-inflammatory and chemotactic cytokines interleukin 1β (IL-1β), IL-6, and RANTES have been assessed by enzyme linked immunoabsorbent assay.25 All of 18 patients studied had increased concentrations of IL-6 and RANTES during active disease compared with healthy controls, and concentrations parallelled disease activity. These cytokines correlated with the ESR but not with CRP values. This lack of CRP correlation (CRP being driven by IL-1 and IL-6) was not adequately explained. The positive correlation with disease activity suggested that these cytokines may contribute to the vasculitis and raised the possibility of their use in monitoring disease and treatment. However, serum cytokine assays are not necessarily a reflection of tissue cytokine concentrations and may not accurately detect biologically active cytokine. Their use over and above the ESR remains to be established.
TREATMENT OPTIONS
Medical treatment
Steroids have formed the mainstay of treatment for Takayasu arteritis and reports of efficacy vary. This may relate to the stage of disease at which treatment is introduced in addition to disease extent. Early data suggested little benefit,1 with six of eight patients treated showing no improvement. Data from the USA in 19856 from 29 steroid treated patients demonstrated a reduction in ESR, a reduction of inflammatory symptoms, and eight of 16 patients with absent pulses were shown to have a return of a pulse after a delay of several months. In a later study, of 48 treated patients, remission was achieved at least once with steroids alone in 60%.10
It is now accepted that approximately half of patients treated with steroids will respond.8 This lack of universal success and the side effects associated with steroid use have led to a search for a more effective treatment.
Comparisons have been made with the treatment of other systemic vasculitides, such as Wegener's granulomatosis.26 Therefore, immunosuppressive agents including cyclophosphamide, azathioprine, and methotrexate have all been tried. However, the difficulty of comparing Takayasu arteritis with Wegener's granulomatosis relates not only to the size of vessel affected by the disease process, but also to the very different morbidity and mortality associated with these disorders. Untreated systemic Wegener's granulomatosis has a mean survival from disease onset of five months and a one year mortality of 82%,27 which is in sharp contrast to that of Takayasu arteritis.
Kerr et al studied 25 steroid unresponsive patients10 receiving cytotoxic medications including cyclophosphamide, azathioprine, or methotrexate, although not concurrently. The overall remission rate was 33%. Twenty three per cent of all treated patients in their study never achieved remission.
Because no single cytotoxic drug appears to be better than any other in terms of efficacy, side effect profiles have been an important driving force in determining treatment. An early report of methotrexate28 suggested that it was a clinically useful, well tolerated drug. A follow up study of 16 steroid unresponsive patients treated with methotrexate and steroid demonstrated remission in 81%.29 However, seven of 16 relapsed as they were weaned off of steroids. Overall, eight patients sustained remissions of four to 34 months and four of these were able to discontinue treatment altogether. Three of 16 progressed despite treatment. A Brazilian study included 12 patients treated with methotrexate and prednisolone13; 58% had a good response. Three had to discontinue treatment because of leucopenia or abnormal liver function.
“Because no single cytotoxic drug appears to be better than any other in terms of efficacy, side effect profiles have been an important driving force in determining treatment”
More recently, three patients have been reported after treatment with mycophenolate mofetil.30 All three showed clinical benefit, steroids were tapered or discontinued, and no toxicity was observed. Larger studies will be necessary to confirm these findings and establish the place of this drug in the treatment of Takayasu arteritis.
Currently, the best evidence based treatments include steroids, to which 50% respond, and methotrexate to which a further 50% respond. The use of methotrexate as a steroid sparing drug is logical and safe. Twenty five percent of patients with active disease will not respond to current treatments and care should be taken not to expose these patients to the hazards of prolonged immunosuppression in the absence of clinical benefit.
The other important medical issues relate to the management of hypertension and the prevention and treatment of thrombosis. Hypertension can be particularly difficult, and worsened by the use of steroids with their fluid retaining side effects. The use of angiotensin converting enzyme inhibitors requires careful monitoring in view of the frequency of renal artery stenosis.31
Surgical treatment
Indications for surgery include hypertension with critical renal artery stenosis, extremity claudication limiting activities of daily living, cerebrovascular ischaemia or critical stenoses of three or more cerebral vessels, moderate aortic regurgitation, and cardiac ischaemia with confirmed coronary artery involvement.10 In general, surgery is recommended at a time of quiescent disease to avoid complications, which include restenosis, anastamotic failure, thrombosis, haemorrhage, and infection.6,10
Surgery may be unnecessary for aortic arch and splanchnic disease as a result of extensive collateral development.31 However, recent surgical experience of critical thoracic aortic arch stenoses and stroke risk from the National Institutes of Health, USA32,33 concluded that critical stenoses should be corrected to prevent stroke, with grafts originating from the ascending aorta. Renal artery involvement is best treated by percutaneous transluminal angioplasty.33 Stent placement following angioplasty for ostial lesions, long segment lesions, incomplete relief of stenoses, and dissection is safe and effective.34 Radical surgical treatment of thoracic aneurysms is recommended if technically possible because more palliative procedures fail to prevent recurrent aneurysm formation or to minimise risk of later surgery.35
PREGNANCY
Because Takayasu arteritis predominantly affects women of reproductive age, the issue of pregnancy is important. Kerr et al reported five pregnancies in their series of 60 patients, all of whom had normal deliveries of a normal live infant.10 Only one patient had disease exacerbation during pregnancy.
A study from Hong Kong in 198336 reported on 13 women who had experienced a total of 30 pregnancies. Apart from hypertension, there were no major obstetric problems and no maternal deaths directly related to pregnancy. Fetal outcome could be predicted on the basis of maternal vessel involvement (abdominal aorta and renal), severity of maternal hypertension, superimposed pre-eclampsia, and timing of adequate blood pressure control.
Maternal complications reported in 12 patients from India37 included superimposed pre-eclampsia, congestive cardiac failure, progressive renal impairment, and one case of postpartum sepsis. Abdominal aortic involvement and a delay in seeking medical attention predicted a poor perinatal outcome.
Fertility is not adversely affected, pregnancy per se does not appear to exacerbate the disease, but management of hypertension is essential. Hypertension in the second stage of labour is a risk factor for cerebral haemorrhage; shortening this stage by use of low forceps delivery or vacuum extraction appears to be a reasonable solution.36,37
LONG TERM FOLLOW UP
Takayasu arteritis is a systemic vasculopathy that can progress to cause vital organ ischaemia. Therefore, long term follow up is recommended. The limitations of monitoring the acute phase response have been discussed; better tools are required and so far these have focused on vascular imaging techniques, with non-invasive methods obviously being most appropriate.
Doppler ultrasound is easily applied to extracranial vessels and can determine vessel wall thickness. Magnetic resonance angiography (MRA) is now being investigated in the evaluation of large vessel vasculitides.38 It provides high resolution detail of vessel wall thickness and lumen configuration, and allows the measurement of wall enhancement as a reflection of oedema and active inflammation. Compared with the gold standard of conventional angiography, approximately 2% of stenosed arteries are overestimated as occluded on MRA. The reduction of enhancement on follow up is presumed to reflect reduced inflammatory activity. Therefore, MRA is likely to be used increasingly as an accurate follow up tool.
The management of patients with Takayasu arteritis can be problematic. There may be uncertainty with regard to the onset and course of the disease, a poor correlation between clinical assessment and disease activity, poor disease activity markers in peripheral blood, and a lack of useful treatment in up to 25% of patients with progressive disease. The risk of increased morbidity and mortality means that most patients who present will ultimately receive immunosuppression. The vasculitides, particularly those affecting small vessels, generally require aggressive treatment. The same may not be true of all patients with Takayasu arteritis despite the angiographic appearances. Cohort studies suggest a good prognosis for those with uncomplicated or monocomplicated disease. Thus, the temptation to immunosuppress such patients aggressively should be questioned. In contrast, early treatment of those with progressive complicated disease may lead to a better prognosis for this group. Because inflammation is a risk factor for atherosclerosis,2 more atherosclerotic complications are likely in the longer term.
Take home messages
-
Takayasu arteritis is rare, affects mainly women, and is most commonly seen in Japan, South East Asia, India, and Mexico, where it usually presents in the 2nd or 3rd decade of life
-
Manifestations range from asymptomatic disease, found as a result of impalpable pulses or bruits, to catastrophic neurological impairment
-
Disease presentation varies between different populations
-
Angiography remains the gold standard for diagnosis
-
The four most important complications for classification are Takayasu retinopathy, secondary hypertension, aortic regurgitation, and aneurysm formation, each being graded as mild/moderate or severe at the time of diagnosis
-
Four grades of disease are described, which can be used for prognostic and treatment assessment: cohort studies suggest a good prognosis for those with uncomplicated or monocomplicated disease
-
Approximately half of those patients treated with steroids will respond, and half of the remaining patients respond to methotrexate; mycophenolate mofetil may be useful
-
Treatment should aim to control disease activity and preserve vascular competence, with minimal long term side effects; those with disease that carries a good prognosis should not be put at risk by treatment that is more harmful than the disease itself
-
Fertility is not adversely affected and pregnancy does not appear to exacerbate the disease, although management of hypertension is essential
“Takayasu arteritis is a systemic vasculopathy that can progress to cause vital organ ischaemia”
As with any rare disorder, sufficient patient numbers for randomised controlled treatment trials are lacking. The aim of treatment must be the control of disease activity and the preservation of vascular competence, with minimal long term side effects. Patients with disease that carries a good prognosis should not be put at risk by treatment that is more harmful than the disease itself. Current evidence favours the use of steroids and methotrexate, but mycophenolate mofetil may prove to have a role.
Acknowledgments
The authors would like to thank Dr M Thornton, Consultant Radiologist, Southmead Hospital, Westbury on Trym, Bristol for help in provision of the radiographic material.