Article Text

Abstract
Since its discovery in 1997, knowledge about theSHOX gene has rapidly increased. In this review, we summarise clinical features and diagnostic and therapeutic implications in SHOX haploinsufficiency and overdosage. SHOX haploinsufficiency usually results in mesomelic short stature and Turner skeletal features, including Madelung deformity with puberty, in subjects with normal gonadal function. Thus, identification of early or mild signs of Madelung deformity is pivotal for the diagnosis, and gonadal suppression therapy may serve to mitigate the clinical features. By contrast, SHOX overdosage usually leads to long limbs and tall stature resulting from continued growth into the late teens in subjects with gonadal dysgenesis. Thus, the combination of tall stature and poor pubertal development is the key to diagnosis, and oestrogen therapy can help the prevention of unfavourably tall stature as well as the induction of sexual development. These findings, in conjunction with skeletal assessment in Turner syndrome and expression analysis during human embryogenesis, imply thatSHOX functions as a repressor for growth plate fusion and skeletal maturation in the distal limbs and, thus, counteracts the skeletal maturing effects of oestrogens.
- SHOX
- oestrogens
- clinical features
- practical implications
Statistics from Altmetric.com
The distal ends of Xp and Yp are composed of 2.6 Mb of DNA sequences that are identical in the X and Y chromosomes.1This particular region is named the short arm pseudoautosomal region (PAR1), where the X and the Y chromosomes recombine during male meiosis.1 Genes on the PAR1 escape X inactivation and, therefore, are present in two active copies in both males and females.1 Since Xp terminal deletions invariably result in short stature irrespective of the breakpoints2 and small Yp terminal deletions lead to short stature,3 it has been suggested that a growth gene resides in the PAR1, and that haploinsufficiency of the growth gene causes short stature as a dominant phenotype.2
In 1997, Rao et al 4 cloned a novel gene from the distal part of the PAR1 and named itSHOX for short stature homeobox containing gene. This gene was also identified by Ellison et al 5 and termed PHOG for pseudoautosomal homeobox containing osteogenic gene.SHOX is expressed from an inactive X chromosome as well as an active X and a normal Y chromosome, indicating that SHOX exerts the dosage effect in sex chromosome aberrations, as expected.4 In addition,SHOX is most strongly expressed in bone marrow fibroblasts, implying that SHOX has a certain role in bone growth and maturation (however,SHOX expression has not been examined in several critical tissues such as chondrocytes or osteocytes).4 ,5 Furthermore, expression analysis in human embryos has shown that SHOX is exclusively expressed in the developing limbs and in the first and second pharyngeal arches, suggesting that it is involved in mesomelic short stature and skeletal features in Turner syndrome.6
In this review, we summarise clinical features and diagnostic and therapeutic implications in SHOXhaploinsufficiency and overdosage.
SHOX haploinsufficiency
PATIENTS
SHOX haploinsufficiency is common to sex chromosome aberrations associated with terminal Xp or Yp deletions. To define the clinical features in SHOXhaploinsufficiency, however, it is necessary to identify patients with intragenic SHOX mutations or microdeletions involving SHOX as the sole impaired disease gene. To date, such mutations or microdeletions have been shown in more than 100 patients, including 29 Japanese patients in whom sufficient clinical data are available4 ,6-11 (unpublished data). The Japanese patients are familial in 17 cases from seven pedigrees and sporadic in 12 cases, and consist of three prepubertal boys, seven adult males, eight prepubertal girls, five pubertal girls, and six adult females. All patients with proven SHOXhaploinsufficiency have low-normal to severe short stature and lack demonstrable non-skeletal Turner features or definitive biochemical abnormalities. In addition, all pubertal or adult patients have normal gonadal function.
SKELETAL FEATURES
Skeletal features are highly variable but can be classified into several groups, on the basis of the combination of short fourth metacarpals and/or cubitus valgus, which appears in ∼40% of Turner patients, and Madelung deformity and/or mesomelia characteristic of Léri-Weill dyschondrosteosis (LWD), which occurs in ∼7% of Turner patients.10 ,12 For example, the 29 Japanese patients are divided into the following: (1) five patients with no discernible abnormalities; (2) one patient with short fourth metacarpals and borderline cubitus valgus but without demonstrable Madelung deformity or mesomelia; (3) three patients with Madelung deformity and/or mesomelia of various degrees but without recognisable short fourth metacarpals or cubitus valgus; and (4) 20 patients with both short fourth metacarpals and/or cubitus valgus and Madelung deformity and/or mesomelia of various degrees (fig 1). In addition, genu valgum and relatively short lower limbs are clinically discernible in all patients with overt LWD, and tibial exostosis was detected in one of four patients with LWD with radiographs of the knees and lower legs. These findings indicate that SHOXhaploinsufficiency causes not only mesomelic short stature but also Turner skeletal features in the limbs,10 and that most patients with SHOX haploinsufficiency have features of LWD to a variable extent, although there may be an ascertainment bias in that patients with the LWD phenotype are predominantly studied for SHOXhaploinsufficiency.

Radiographs of the hands and wrists in patients with proven SHOX haploinsufficiency and normal gonadal function. (A) Normal skeletal appearance in a 15 year old boy. (B) Short fourth metacarpal in a 14 year old girl. (C) Moderate Madelung deformity in a 13 year old girl. (D) Short fourth metacarpal and severe Madelung deformity in an 18 year old girl.
Limb skeletal features are more severe in females than in males and become evident with puberty.10 ,13-15 In this context, two matters are noteworthy. First, short fourth metacarpals in Turner syndrome are often associated with radiologically discernible premature fusion of the epiphysis,15 ,16 and Madelung deformity is primarily ascribed to premature fusion of the medial half of the distal radial and ulnar growth plates.17 In addition, bone maturation in Turner syndrome is relatively advanced in metacarpal and distal radioulnar regions.18 These findings, in conjunction with SHOXexpression in the developing limbs,6 suggest thatSHOX functions as a repressor for growth plate fusion and skeletal maturation in the distal limbs, so thatSHOX haploinsufficiency results in premature growth plate fusion and relatively advanced skeletal maturation in such regions. Second, skeletal maturation in normal subjects is primarily caused by gonadal oestrogens in both sexes,19 and serum oestrogen concentration is higher in females than in males and increases with puberty in both sexes.20 Thus, it has been suggested that gonadal oestrogens exert a maturational effect on skeletal tissues that are susceptible to unbalanced premature growth plate fusion and skeletal maturation because ofSHOX haploinsufficiency, facilitating the development of skeletal lesions in a female dominant and pubertal tempo influenced fashion.10 In agreement with this, severe skeletal features are usually manifested by early maturing females who are exposed to gonadal oestrogens from a relatively early age.10 Furthermore, this notion would explain why the prevalence of skeletal lesions in Turner syndrome remains roughly 40% for short metacarpal and cubitus valgus and only about 7% for Madelung deformity, because gonadal oestrogen production is usually severely compromised in Turner syndrome.10 Although Turner patients treated with oestrogens rarely have Madelung deformity, this would be because of oestrogen therapy usually being started from the late teens with a low dosage.21 However, a hitherto unknown modifying factor(s) other than oestrogens would also be relevant to the development of skeletal lesions, because skeletal features are still variable among age and sex matched patients.
In Turner syndrome, skeletal anomalies also frequently appear in the faciocervical region.12 In this context, the presence of high arched palate in two adult Japanese females and short neck in three German patients6 ,10 (unpublished data) together with SHOX expression in the first and second pharyngeal arches,6 suggests thatSHOX haploinsufficiency is also relevant to Turner skeletal features in the faciocervical region. However, since the prevalence of faciocervical skeletal anomalies is apparently low as compared with that of limb skeletal anomalies, it appears that, in Turner syndrome, a deformational effect of cystic hygroma and facial oedema on the developing skeletal tissues also contributes to the development of faciocervical skeletal features.10
GROWTH PATTERN
Prenatal growth appears to be compromised inSHOX haploinsufficiency. The mean SD score is –1.1 for birth length and –0.6 for birth weight.10Since the mean SD score is –1.01 for birth length and –1.20 for birth weight in Turner syndrome,22 SHOX haploinsufficiency may account for most of the birth length reduction and roughly half of the birth weight reduction in 45,X Turner syndrome.
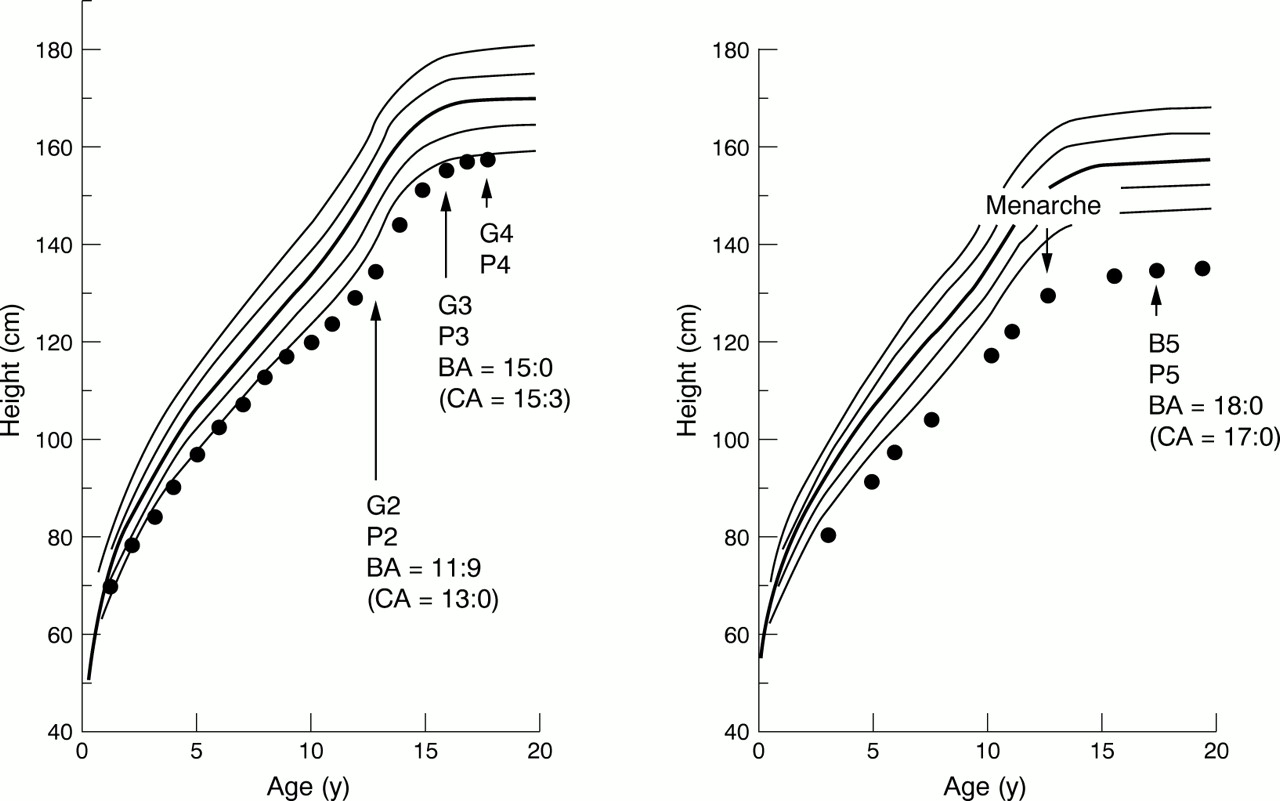
Postnatal growth primarily depends on the presence or absence of overt LWD. Most patients without overt LWD continue to grow along the –2 SD growth curve throughout the growth period (fig 2).10 The magnitude of height deficit (about 2 SD) is compatible with the estimation that SHOX haploinsufficiency decreases adult height by about 12 cm in the absence of overt LWD,23 because 12 cm approximates the magnitude of 2 SD of the adult height in the normal population. Consistent with the relatively mild statural effect of SHOXhaploinsufficiency (about 2 SD) as compared with the magnitude of normal height variation (±2 SD, that is, 4 SD),SHOX haploinsufficiency permits normal height in patients with a high genetic height potential (tall parental height).10 Furthermore, the growth pattern with an apparently normal growth rate, which is similar to that of normal variant short stature, appears to be characteristic ofSHOX haploinsufficiency, because most diseases affecting linear growth are usually associated with a reduced growth rate.24

Growth charts in patients with proven SHOX haploinsufficiency and normal gonadal function.10 The actual heights are plotted on the sex matched standard longitudinal growth curves (mean, ±1 SD, and ±2 SD) for Japanese children. Pubertal stage is indicated according to the classification of Tanner (G: genitalia; B: breast; P: pubic hair). Bone ages (BAs) are given together with chronological ages (CAs) at the time of BA determination. (Left) A boy without recognisable skeletal abnormalities (see fig 1A). (Right) A girl with both short fourth metacarpal and Madelung deformity (see fig 1D).
By contrast, patients with overt LWD, though they usually grow along the –2 SD growth curves before puberty, show an obvious downward growth shift with puberty (fig 2).10 It is likely that prepubertal growth is relatively well preserved because of dormant gonadal function, whereas pubertal growth is compromised because of production of gonadal oestrogens facilitating growth plate fusion.
SHOX haploinsufficiency alone is unlikely to explain the growth failure and the growth pattern in 45,X Turner syndrome.2 In 45,X Turner syndrome, the mean adult height is about 20 cm lower than that of normal females, and the linear growth is accompanied by a reduced growth rate beginning from early childhood in the absence of discernible LWD.2 ,22 Here, it is noteworthy that 45,X is associated with gross chromosome imbalance, because chromosome imbalance, irrespective of the increase or decrease of chromosomal material, has been suggested to result in global developmental defects including growth failure.25 Although 45,X Turner patients usually have gonadal dysgenesis, gonadal oestrogen deficiency is unlikely to influence adult height or childhood growth pattern.2 ,26 Thus, the remaining growth deficit and the reduced growth rate from early childhood in 45,X Turner syndrome appears to be the result of chromosome imbalance.
DIAGNOSTIC IMPLICATIONS
The above clinical findings provide some indications for the selection of patients to be examined forSHOX haploinsufficiency. First, skeletal assessment implies that SHOXhaploinsufficiency is usually detected in patients with the LWD phenotype and is obviously rare in patients with no skeletal abnormalities or in those with short metacarpals and/or cubitus valgus only. This would be further supported by the following findings: (1) of the five Japanese patients with no skeletal abnormalities, three are prepubertal children who may manifest skeletal features with puberty, so that definitive normal skeletal findings are manifested by only two adult males with borderline short stature ascertained through familial study of the probands with Madelung deformity; (2) we could identify a single prepubertal girl with SHOX mutation and she began to show mild Madelung deformity with puberty (unpublished data); (3) although Rao et al 4 found a family with a heterozygousSHOX nonsense mutation by analysing 91 patients with short stature, and Binder et al 11 found a girl withSHOX deletion by studying 68 patients with short stature, re-examination of such patients has shown mild skeletal features in most of them6 ,11; and (4) we have not identified SHOX haploinsufficiency in eight subjects with short stature who exhibit short metacarpals and/or cubitus valgus but lack Madelung deformity (unpublished data). Second, growth assessment suggests that most patients withSHOX haploinsufficiency usually have borderline short stature (around –2 SD) and grow along the standard growth curves before puberty. These findings imply thatSHOX haploinsufficiency should primarily be looked for in patients with Madelung deformity and/or mesomelia, and that the proband's family members should also be examined if they have no discernible skeletal features or short stature. By contrast,SHOX haploinsufficiency is quite unlikely in patients with no evidence of Madelung deformity or mesomelia, especially in those with severe short stature (<3 SD) accompanied by a reduced height velocity.
For the detection of SHOXhaploinsufficiency, therefore, it is critical to identify characteristic skeletal features. In particular, it is of practical importance to recognise early or mild signs of Madelung deformity on hand and wrist radiographs that are almost invariably obtained for bone age evaluation in children with short stature. For this purpose, it is necessary to observe carefully several indications of Madelung deformity such as metaphyseal lucency and epiphyseal hypoplasia at the ulnar border of the distal radius, decreased carpal angle, angulation of the distal radius and ulna, and subluxation of the distal ulna.27 In our experience, the first signs of Madelung deformity are often exhibited by metaphyseal lucency and epiphyseal hypoplasia of the medial side of the distal radius in prepubertal patients and by decreased carpal angle in pubertal or adult patients (fig 3). When such findings are suspected, radiographs of the distal limbs should be obtained, to examine characteristic features such as radial curvature and/or shortening and tibial deformity.

Early and mild signs of Madelung deformity. (A) Metaphyseal lucency and mild epiphyseal hypoplasia of the medial side of the distal radius in an 8 year old boy. (B) Decreased carpal angle in addition to angulation of the distal radius and epiphyseal hypoplasia of the medial side of the distal radius in a 15 year old girl.
In the analysis of SHOX haploinsufficiency, it is noteworthy that microdeletions involvingSHOX are much more prevalent than intragenicSHOX mutations. For example, of 29 Japanese patients, 28 patients have microdeltions, and only a single patient has an intragenic mutation10 (unpublished data) and similar data have also been published.7-9 ,11 The high prevalence of microdeletions would be consistent with repetitive sequences such as subtelomeric interspersed repeats being abundantly present aroundSHOX,28 because unequal crossing over between homologous chromosomes or an intrachromosomal recombination is prone to occur in such a region. Thus, it is recommended that FISH analysis should be performed first and whenSHOX deletion is excluded intragenic mutation should be searched for.
THERAPEUTIC IMPLICATIONS
For SHOX haploinsufficiency, two therapeutic interventions can be considered. First, since growth hormone (GH) therapy is effective in Turner syndrome despite the absence of GH deficiency,29 it may also be advantageous inSHOX haploinsufficiency. To date, beneficial short term effects have been reported in three patients.11 ,30 Second, since gonadotrophin releasing hormone analogue (GnRHa) therapy can suppress gonadal oestrogen production, it may serve to prevent or mitigate the development of skeletal features. The assessment of GH and GnRHa therapy awaits further and future clinical studies.
SHOX overdosage
PATIENTS
SHOX overdosage can be caused by structural, as well as numerical, abnormalities of the sex chromosomes. In this context, females with 46,X,idic(Xq–) (=46,X,der(X)t(Xq;Xq))31–34 (unpublished data) and 46,X,der(X)(pter→q21::p11→pter) (=46,X,der(X)t(Xp;Xq))35-37 are noteworthy, because they have a unique combination of SHOX overdosage (one on the normal X chromosome and two on both ends of the derivative X chromosome) and oestrogen deficiency owing to gonadal dysgenesis. This combination shows a sharp contrast to the association ofSHOX haploinsufficiency with normal gonadal oestrogen production.
SKELETAL FEATURES
Skeletal data are fragmentary in each individual patient. Taken together, however, it is likely that the combination ofSHOX overdosage and oestrogen deficiency leads to long metacarpals, large hands and feet, and long limbs31-37 (unpublished data). SinceSHOX is expressed in the distal limbs, this phenotype can be explained by the SHOXdosage effect which could be exaggerated because of the delayed growth plate fusion resulting from gonadal oestrogen deficiency. This idea would also explain why relative tall stature in Klinefelter patients is almost totally the result of increased leg length,38because Klinefelter patients have three copies ofSHOX and hypogonadism.
GROWTH PATTERN
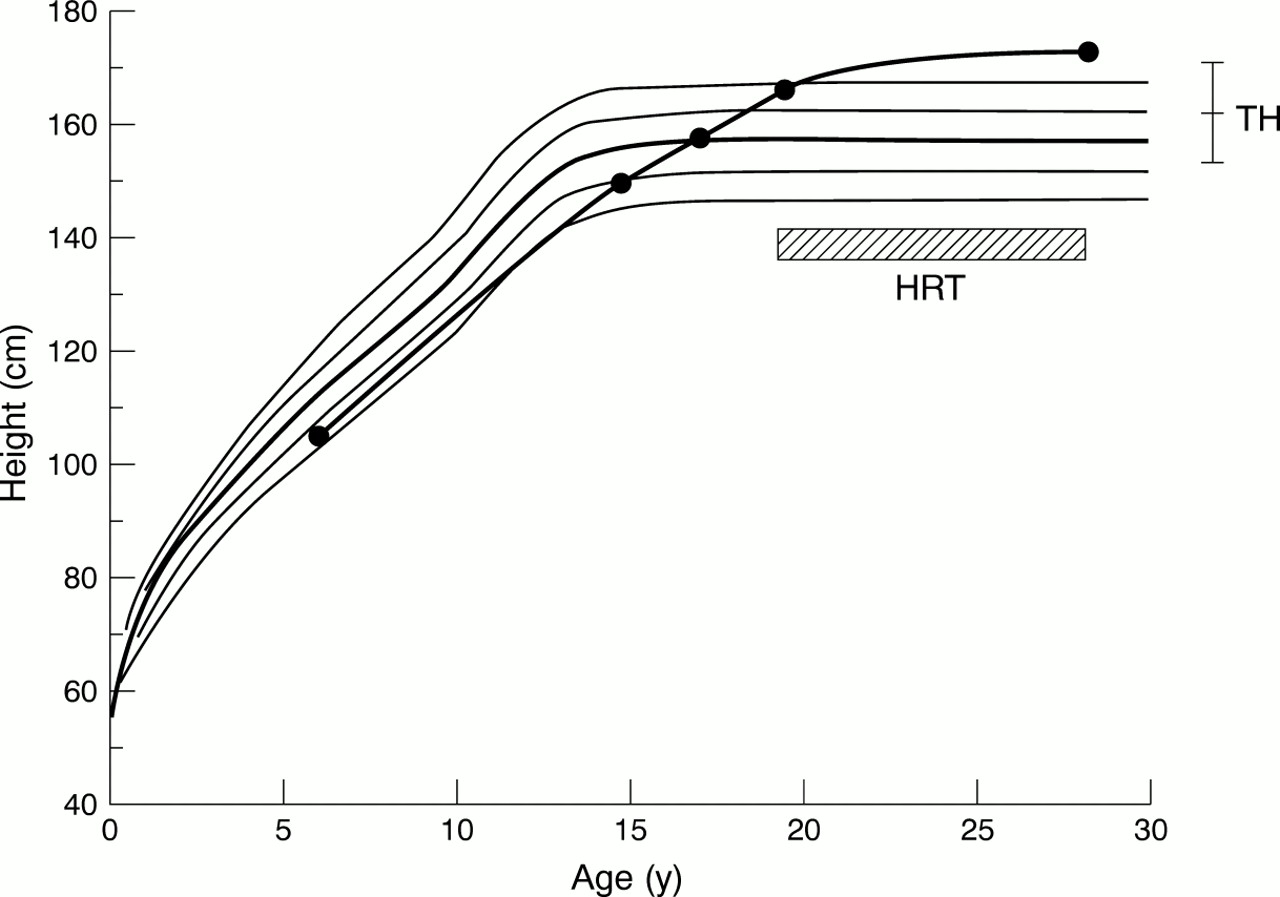
Long term growth pattern is available in only a few patients36 ,37 (unpublished data). Collectively, they grow along the normal height standard curves in the prepubertal period and continue to grow with a nearly constant height velocity throughout the pubertal period of normal subjects, attaining tall adult height (above +2 SD) (fig 4). The white adult height is 174.8 (SD 4.6 cm) (n=5) for females with apparently non-mosaic 46,X,idic(Xq–) and primary amenorrhoea untreated until their late teens (over 17 years old).36
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Growth curve in a female with 46,X,der(X)(pter→q21::p11→pter) accompanied by proven SHOX overdosage and complete gonadal dysgenesis.36 The growth curve of the female is depicted on the standard growth curves for Japanese females (mean, ±1 SD, and ±2 SD). Target height (TH, a child's final height as predicted from the parental height) is shown, together with target range (95% confidence interval of target height) indicated by the vertical bar. HRT: hormone replacement therapy.
Two factors are relevant to the tall stature. One is theSHOX duplication on the derivative X chromosome, because SHOX has a dosage effect on the adult height.2 The other is gonadal dysgenesis, because gonadal oestrogen deficiency permits a prolonged growth period. Each factor alone appears to be insufficient to explain the tall stature. For example, the white adult height is 167.9 (SD 7.7) cm (n=19) for females with 47,XXX accompanied by three copies ofSHOX and relatively well preserved oestrogen production, 164.3 (SD 7.7 cm) (n=22) for females with 46,XX gonadal dysgenesis accompanied by two copies of SHOXand gonadal oestrogen deficiency, and 162.2 (SD 6.0) cm for normal females accompanied by two copies ofSHOX and gonadal oestrogen production.2 Thus, the tall stature in females with 46,X,idic(Xq–) and 46,X,der(X)(pter→q21::p11→pter) appears to be the result of the synergistic effects of the two factors.36
The growth pattern is similar to that in oestrogen resistance or aromatase deficiency, which is free from biological effects of both gonadal and extragonadal oestrogens.18 This suggests that an extra copy of SHOX has the potential to override the skeletal maturation effect of extragonadal oestrogens, because SHOX is likely to function as a repressor for skeletal maturation.
DIAGNOSTIC IMPLICATIONS
SHOX overdosage should be looked for in females with tall stature and gonadal dysgenesis, especially in those with continued growth into the late teens. In addition, large hands and feet and long limbs represent a further indication forSHOX overdosage.
THERAPEUTIC IMPLICATIONS
There are two therapeutic implications. First, oestrogen replacement therapy is effective in preventing unfavourable tall stature as well as inducing pubertal development. Second, although GH therapy has widely been used in Turner syndrome,29 it may be unnecessary in females with the unique combination.
Summary
Current data indicate that there is a striking contrast in clinical features between patients with the association ofSHOX haploinsufficiency with normal gonadal function and those with the combination ofSHOX overdosage and gonadal dysgenesis. This is consistent with the notion that SHOXfunctions as a repressor for growth plate fusion and skeletal maturation in the distal limbs and counteracts the skeletal maturing effects of oestrogens. Further clinical studies in patients withSHOX abnormalities will permit a better definition of the phenotypic spectrum and better diagnostic and therapeutic applications.
Acknowledgments
SHOX sequence analysis was carried out in collaboration with Dr Rappold, Institute of Human Genetics, Heidelberg University, Germany.