Article Text

Abstract
Three infants, who presented with intestinal obstruction due to diffuse transmural intestinal ganglioneuromatosis, are described. Mutation analysis of exon 16 of the RETproto-oncogene revealed germline M918T and thus, a molecular diagnosis of multiple endocrine neoplasia type 2B (MEN 2B). Two infants developed medullary carcinoma of the thyroid. The third had a prophylactic thyroidectomy despite no obvious thyroid masses and normal calcitonin concentrations, but microscopic multifocal medullary carcinoma was found on histological examination. Early recognition of intestinal ganglioneuromatosis with germlineRET M918T mutation in pseudo-Hirschsprung’s disease is an indication for prophylactic thyroidectomy.
- intestinal ganglioneuromatosis
- RET
- MEN 2B
- thyroidectomy
Abbreviations used in this paper
- IND
- intestinal neuronal dysplasia
- MEN
- multiple endocrine neoplasia
- NGF
- nerve growth factor
- MTC
- medullary thyroid carcinoma
- ERK
- extracellular signal related kinases
Statistics from Altmetric.com
Functional intestinal obstruction in infancy is commonly due to Hirschsprung’s disease but may also be due to a variety of other enteric neuromuscular disorders. Here, we report three infants with transmural intestinal ganglioneuromatosis who presented in a Hirschsprung-like manner. The RETproto-oncogene is a susceptibility gene for both Hirschsprung’s disease and multiple endocrine neoplasia (MEN) type 2.1 In general, gain of function RET mutations are associated with MEN 2 and loss of function mutations with Hirschsprung’s disease. In these infants we describe their presentation, course, histological findings,RET mutation analysis, and the implications of this for their management.
Case histories
CASE 1
A female infant was born at term by normal delivery. She presented at two weeks of age with intestinal obstruction. A full thickness rectal biopsy revealed extensive ganglioneuromatosis of the myenteric and submucosal plexuses. The obstruction was initially relieved by a colostomy but later required conversion to an ileostomy. Full thickness samples of the colon and the small bowel showed extensive ganglioneuromatosis. Germline mutation analysis revealed the presence of de novo M918T of the RET gene in the patient. The diagnosis of MEN 2B was made. At three years of age, she was found to have short stature, frontal bossing, and notable lumbar lordosis. However, no specific diagnosis of a skeletal dysplasia could be made. There was no evidence of medullary carcinoma of the thyroid (MTC) or phaeochromocytoma on repeated ultrasound scanning of thyroid and abdomen, urine vanillylmandelic acid and catecholamine excretion, and pentagastrin stimulated calcitonin concentrations. One year later, at the age of four, a routine pentagastrin stimulated calcitonin was elevated and a total thyroidectomy was carried out. Histopathological examination revealed two nodules of MTC. A total colectomy was performed at five years of age and the entire colon and terminal ileum showed extensive transmural ganglioneuromatosis.
CASE 2
A male infant first presented at the age of 15 months with severe constipation since birth, coarse facies, gross motor delay with myopathic electromyography, and unusual fat distribution. He had a large suprapubic pad and decreased fat around the pelvis. Mucopolysaccharidoses were excluded. A muscle biopsy was mildly myopathic with no clear diagnostic features. The severe constipation was considered to be secondary to the neurological problems and was not investigated further at that time. At the age of four years he presented with thyroid nodules; a thyroidectomy was performed, and on histopathology, MTC was diagnosed. Germline mutation analysis revealed de novo RET M918T. Subsequently, the diagnosis of intestinal ganglioneuromatosis was made on a full thickness biopsy, an ileostomy was raised, and the patient underwent total colectomy. The entire resected colon and terminal ileum showed extensive florid transmural ganglioneuromatosis.
CASE 3
A male infant was born at term by normal delivery. He presented with enterocolitis and large bowel obstruction at the age of 13 days. At another institution, Hirschsprung’s disease was excluded on a rectal biopsy. The diagnosis of “intestinal neuronal dysplasia” (IND) was suggested and a defunctioning colostomy was raised. At the age of three years, a repeat rectal biopsy and biopsies taken distal and proximal to the stoma all showed diffuse transmural ganglioneuromatosis and an increase in acetylcholinesterase positive fibres in the lamina propria. Repeated ultrasound scanning of thyroid and abdomen together with urinary catecholamines and pentagastrin stimulated calcitonin concentrations failed to reveal evidence of MTC or phaeochromocytoma. Germline mutation analysis showed the presence of de novo RET M918T. At six years of age, ileorectal pull through was successfully carried out. The resected colon and terminal ileum both showed extensive ganglioneuromatosis in the submucosal and myenteric plexuses. Six months later prophylactic thyroidectomy was performed, although no obvious thyroid mass was seen on ultrasound scans, and serial pentagastrin stimulated calcitonin concentrations were normal. In addition, no adrenal abnormality was seen on imaging and repeated 24 hour urine samples for catecholamines were consistently normal. Histopathological examination of the resected thyroid specimen revealed microscopic multifocal MTC.
Discussion
Multiple endocrine neoplasia type 2B syndrome is a serious condition which almost always initially presents with severe constipation, diarrhoea where there is enterocolitis, and even frank obstruction, often in infancy.2 ,3 In many cases, other external stigmata of MEN 2B such as a characteristic facies, “blubbery lips” from mucosal neuromas, marfanoid habitus, medullated corneal nerve fibres, and even MTC, are already present.2 ,4 Indeed, in virtually 100% of MEN 2B patients, medullary carcinoma of the thyroid, a potentially lethal tumour, eventually occurs. This tumour can only be cured with surgery4; early detection is therefore crucial as MTC responds poorly to radiotherapy and chemotherapy. As MTC is of neuroendocrine, and not follicular epithelial origin, radioiodine therapy, which can be efficacious in metastatic follicular thyroid carcinoma, has no role in metastatic MTC.
All three infants presented with a pseudo-Hirschsprung’s condition for which there are a number of candidate disorders. In MEN 2B intestinal obstruction and severe constipation are associated with transmural intestinal ganglioneuromatosis; it is thus imperative to recognise the morphological appearances of ganglioneuroma. These are massive proliferations of neural tissue (neurones, supporting cells, and nerve fibres) which appear as thickened nerve trunks embedded with mature nerve cells (figs 1 and 2). Ganglioneuroma must not be confused with the diagnosis of IND which includes a number of quite different morphological phenotypes.5 ,6 The diagnosis of IND was erroneously made in one of our patients, thus delaying the correct diagnosis of MEN 2B. It seems that it is the transmural ganglioneuromatosis, particularly involving the myenteric plexus, which is associated with MEN 2B.7 Whereas ganglioneuromata in the lamina propria of the mucosa are seen in von Recklinghausen’s disease this represents a separate entity that does not develop MTC, although 1% of patients with von Recklinghausen’s disease may develop phaeochromocytoma.8
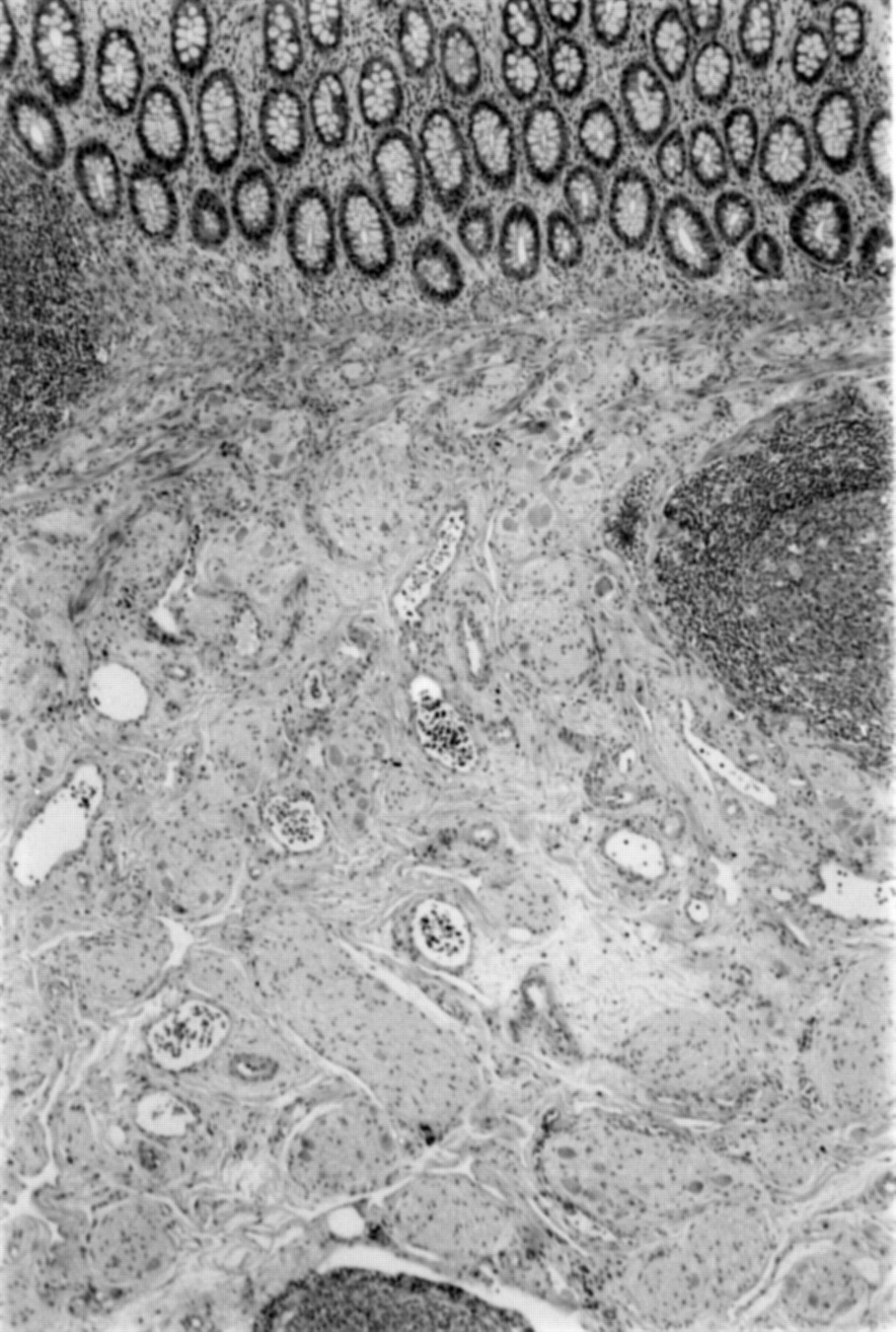
Section of mucosa and submucosa showing extensive ganglioneuromatosis filling the submucosa (case 3). Original magnification × 50.

{kind=link}
{kind=link}
Section showing the myenteric plexus which comprises one giant ganglioneuroma. Note thick nerve trunks embedded with mature neurones (arrowhead). Original magnification × 200.
MEN 2B is a dominantly inherited disorder but at least half of all patients present with a de novo mutation as only few survive to reproductive age or are disabled by a variety of neurological symptoms impairing reproduction. The precise molecular abnormality is in the RET proto-oncogene, which encodes a tyrosine kinase receptor expressed particularly in neural crest derived cells including the enteric ganglia of the gastrointestinal tract. Of patients with MEN 2B 95% have a specific germline point mutation in RET in exon 16 at codon 918 (M918T) and fewer than 4% have a point mutation at codon 883 (A883F).9 ,10 The M918T mutation has been shown to alter RET substrate specificity and seems to act in a ligand independent fashion in the absence of dimerisation although further activation can be achieved in the presence of ligand.11-13 Receptor tyrosine kinases with these mutations also bind to and phosphorylate substrates preferred by non-receptor tyrosine kinases. A883FRET has not been directly tested but given its location, kinase specificity may also be altered in this mutant.10
The receptor is expressed in neural crest cell derived enteric nervous system, adrenal medulla, parathyroid, and C cells of the thyroid. The gain of function mutations are susceptibility factors for endocrine tumours, most notably for medullary carcinoma of the thyroid and to a lesser extent for phaeochromocytoma of the adrenal gland and hyperparathyroidism.
The molecular mechanisms by which RETM918T contributes to the development of neuroendocrine neoplasms and intestinal ganglioneuromatosis remain largely unknown but a number of factors appear clear. PC12 cells (a rat phaeochromocytoma cell line) transfected with M918T mutant RET are unresponsive to nerve growth factor (NGF) induced inhibition of cell proliferation.14 This unresponsiveness is associated with failure of extracellular signal related kinases (ERK) to translocate from the cytoplasm into the nucleus to induce immediate early transcription of C-FOS and Krox 24 despite enzyme activation with NGF. A similar mechanism could result in loss of inhibition of cell proliferation occurring in the thyroid C cells and enteric neurones in patients with MEN 2B. However, other studies have shown that mutatedRET may trigger alternative distinct signalling pathways, including activation of the mitogen activated protein kinase-Jun kinase 115 and paxillin, a CRK associated cytoskeletal protein,16 that could result in transformation of thyroid C cells and enteric neural proliferation.
Our infants all had extensive transmural ganglioneuromatosis and each had a de novo mutation in RET (M918T) and thus a molecular diagnosis of MEN 2B. Case 2 illustrates the necessity for early recognition of an M918T mutation. The signs and symptoms of MEN 2B were not appreciated; the severe constipation was ignored and was not investigated until the child developed MTC. As MEN 2B ultimately and probably inevitably results in MTC, any infant with the diagnosis of intestinal ganglioneuromatosis must have molecular analysis of RET to confirm or exclude germline M918T or A883F. Together these mutations account for more than 97% of MEN 2B cases.9 ,10
Cases 1 and 3 illustrate that when the diagnosis is established by noting transmural intestinal ganglioneuromatosis and confirming the diagnosis of MEN 2B by the highly sensitive and specific molecular test for mutations in RET, the treatment of choice is prophylactic thyroidectomy. Monitoring the calcitonin concentrations and scanning for adrenal and thyroid masses is not sufficient as microscopic MTC can be present without raised calcitonin concentrations, even with pentagastrin stimulation or without identifiable masses on imaging.17 ,18
In all three of our patients transmural ganglioneuromatosis involved the entire colon and the resected terminal ileum, presenting as pseudo-Hirschsprung’s disease. The intestinal symptoms apparently preceded the development of MTC. All had M918T germline mutation ofRET. As surgery is the only curative procedure for MTC we would recommend that all patients who present with pseudo-Hirschsprung’s disease have adequate full thickness biopsies done. Those with transmural intestinal ganglioneuromatosis should undergo molecular diagnostic testing by RETmutation analysis. If germline M918T or A883F mutations are found a prophylactic thyroidectomy should be done. Continued surveillance of the adrenal glands is required with abdominal ultrasound scanning, and urinary catecholamines and vanillylmandelic acid as the patients have at least a 50% chance of developing phaeochromocytoma.
Acknowledgments
This work was undertaken by Great Ormond Street Hospital for Children NHS Trust who received a proportion of its funding from the NHS Executive; the views expressed in this publication are those of the authors and not necessarily those of the NHS Executive.
Abbreviations used in this paper
- IND
- intestinal neuronal dysplasia
- MEN
- multiple endocrine neoplasia
- NGF
- nerve growth factor
- MTC
- medullary thyroid carcinoma
- ERK
- extracellular signal related kinases