





Abstract
Pulmonary alveolar microlithiasis (PAM) is a rare parenchymal lung disease caused by variants in the SCL34A2 gene and characterised by the accumulation of intra-alveolar microliths. PAM has been reported in fewer than 1100 cases throughout the world. It is an autosomal recessive hereditary disease and often associated with consanguinity. Progress with respect to the genetic background and pathophysiology has resulted in an increased understanding of the disease in recent years. Until now, 30 genetic different SLC34A2 variants have been reported, which all are considered significant for disease development. There is no sex difference and the majority of cases are diagnosed at the age of 30–40 years. Many patients are asymptomatic and the diagnosis is made at random. When symptomatic, dyspnoea, cough, chest pain and fatigue are common complaints. The diagnosis of PAM can confidently be based on typical radiographic findings and genetic testing proving rare biallelic SCL34A2 gene variants. Bronchoalveolar lavage and histopathology may show microliths. There is no disease-specific treatment and management is supportive. Lung transplantation should be considered in advanced cases.
Abstract
Pulmonary alveolar microlithiasis is a rare, autosomal recessive lung disease. Formation of alveolar microliths is caused by SCL34A2 variants. The prognosis is severe; respiratory failure is common. Lung transplantation is the only effective therapy. https://bit.ly/3dpkbRw
Introduction
Pulmonary alveolar microlithiasis (PAM) is a rare autosomal recessive lung disease characterised by the accumulation of calcium phosphate deposits (calcospherites or microliths) in the alveoli despite normal serum calcium and phosphorus, and absence of any systemic disease of calcium metabolism [1]. PAM is often diagnosed coincidentally; patients are often asymptomatic until 30–40 years of age. The disease cause is variable. Prognosis may be good, but many patients will develop progressive disease with restrictive lung function impairment, dyspnoea and cough, hypoxia and respiratory failure, secondary pulmonary hypertension and ultimately death. Until today, nearly 1100 cases have been reported in the literature.
The first description of PAM is attributed to Italian biologist and physician, Malpighi [2], who in 1686 in his work “In vesciculis pulmonum innumeri lapilli sunt” describes that “the lungs were heavy and compact with patches of black countless small stones in the interior of the lungs”. In 1918, Harbitz [3] diagnosed the first case in Norway as an autosomal, recessive disease with extensive intra-alveolar microliths on autopsy called Harbitz syndrome and in 1933, Puhr [4] renamed the disease as PAM. In recent years, studies have unveiled the genetic background of PAM, resulting in an improved understanding of the disease, which will hopefully in the future result in better treatment options.
Epidemiology
Until today, a little fewer than 1100 cases of PAM have been reported since Harbitz [3] published the first case in 1918. The majority of patients reported are from Asia and Europe. Turkey, China, Japan, India, Italy and the United States have reported the majority of cases, with the highest number of cases in people of Turkish and Italian descent [5]. Two clusters have been reported, one in Italy and another in Turkey. However, patients with PAM have been identified worldwide [6].
PAM affects both sexes with a slight predominance in males (male/female ratio 0.56) [6]. Almost 40% of cases are diagnosed before the age of 20 years and almost 90% before 50 years, although PAM may be recognised later in life. PAM in children is also reported and a recent study found 28 cases with a median age at first symptoms of 22 months, equivalent to 2–3% of all PAM patients [5, 7–9]. Both sporadic and familial cases are published and a positive family history in some populations is reported in 37% of cases [6]. In families with PAM, the transmission is usually horizontal and if vertical transmission is present, this is exclusively due to consanguinity [6].
Genetic background and pathophysiology
The gene responsible for PAM has been mapped to 4p15.2 and identified as the SLC34A2 gene (solute carrier family 34 member 2) (Entrez Gene ID 10568). SLC34A2 has 13 exons, of which the first is noncoding and the longest transcript encodes a 690 amino acid protein, the sodium-dependent phosphate transport protein 2B (NaPi-2b) (NP_006415). NaPi-2b is expressed in human epithelial cells and in the lungs in alveolar type II cells [10–13].
NaPi-2b is presumed to play an important role in type II alveolar cells with regard to the cell uptake of phosphate from the alveolar fluid [14, 15]. Decreased activity or loss of function of the transporter due to SLC34A2 variants may therefore lead to accumulation of phosphate in the alveolar lumen due to insufficient alveolar phosphate uptake. Excess of phosphate in the alveolar space may lead to phosphate precipitation with calcium in the extracellular fluid and formation of intra-alveolar calcium phosphate stones (i.e. microliths) [16, 17].
To date, 30 different SLC34A2 variants in PAM have been reported in the literature and are located in most parts of the coding regions of the gene [18]. Variants include one or a few nucleotides or deletions involving either a single exon or several exons (table 1). In a recent report, 12 unrelated patients from several European countries and the United States have been reported with 10 different allelic variants in the SLC34A2 gene. In addition, in a Turkish report, seven unrelated PAM patients were found with six different SLC34A2 variants [18, 19]. In one of the patients, variants were present in a compound heterozygous state, whereas a homozygous-variant state was reported in the rest of the patients [18, 19].
SLC34A2 variants reported in pulmonary alveolar microlithiasis
Some authors have pointed out the relationship between the geographical origin and the variant localisation, with patients from China and Japan reported with variants in exon 8 [39]. In addition, the variant c.1402_1404delACC in exon 12 has so far only been reported in patients from Europe [18]. In a recent published cohort of patients, a correlation between phenotype and genotype severity was found [18]. However, it still remains to be investigated whether a genotype–phenotype correlation exists in PAM. Like many other rare autosomal recessive disorders, PAM is associated with consanguinity and a significant part of the patients have at least one sibling who is also affected by the disease [6].
Symptoms and clinical signs
PAM presents most commonly in the second and third decade of life but may be seen at any age [6, 18, 40]. Diagnosis is often made at a more advanced age, presumably because many cases are asymptomatic until then. PAM is a heterogeneous disease and clinical characteristics may range from asymptomatic to pronounced symptoms with right heart and respiratory failure. Disease progression is unpredictable with some patients remaining stable for many years. The clinical hallmark of the disease is the clinical–radiological dissociation, with extensive radiological involvement disproportionate to the subjective complaints, especially in the early phases of the disease [1].
When symptoms are present, the most common complaints are dyspnoea, dry cough, chest pain and fatigue, but also hypoxia, cyanosis, clubbing, haemoptysis, pneumothorax, weight loss and weakness are reported [1, 5, 6, 41]. The dry cough is believed to be cause by the stimulation of unmyelinated c fibres in the bronchial tree and lung parenchyma by microliths [42, 43].
The clinical appearance is often normal but upon progression, fatal respiratory or cardiac failure may develop with accompanying clinical features such as peripheral oedema, liver enlargement, tachypnoea and cyanosis [5, 6].
Extrapulmonary manifestations
Several patients have reported extrapulmonary calcifications such as medullary nephrolithiasis, calcifications in the lumbar sympathetic chain, prostate, aorta and mitral valves, pericardium, the gastric wall, testicles, punctuate calcifications in seminal vesicles and periurethral and epididymal calcifications [6, 18]. The extrapulmonary calcifications can cause symptoms in PAM such as infertility, haematuria and gallstone symptoms.
Some authors have suggested the usage of positron emission tomography (PET) for the identification of extrapulmonary involvement [44]. 18F sodium fluoride PET/computed tomography (CT) based on increased fluoride uptake in hydroxyapatite crystals (microliths), may be the preferred method as it has shown a higher uptake of radiotracer in the lungs, defining a larger extent of pathological involvement compared to 18F-fluoro-2-deoxyglucose (18F-FDG) PET/CT in two PAM patients [44, 45]. 18F-FDG PET/CT has thus been proposed as useful in evaluation of pulmonary inflammation grade. Low 18F-FDG uptake in the lungs has been reported in several patients and may represent a “burned out” inflammatory process [44–47]. Contrary to this, no significant inflammation was found at autopsy in a patient with a high uptake of 18F-FDG in the lungs [48].
PAM in children
Until 2019, 28 published cases of PAM in children younger 6 years of age including 7 infants under 2 years and 2 newborn premature twins have been reported [7]. The majority of the patients (65%) below 5 years of age originated from Asia and 25% of the cohort came from Turkey. The geographical origin and sex distribution were similar to the cohort from Castellana that included all ages of PAM [6, 7].
Symptoms in children with PAM were similar to symptoms in adult PAM patients but the children seem to be more symptomatic and to have more bronchitis and dry cough. Fever, which is not observed in adults, was observed in 30% of the children at presentation. Also contrary to adult patients, some of the children presented with acute respiratory failure [7]. Some speculate that early onset PAM may represent a more severe disease, potentially explained by promoting factors such as genetic and epigenetic factors. Also, environmental factors such as exposure to passive smoking and viral infections may be able to modify the course of the disease. Respiratory infections with cough and fever are common in children in this age group and it cannot be ruled out that PAM was fortuitously diagnosed in an early stage due to symptoms related to viral and other infections.
Three groups of PAM children with PAM might exist: one group with fortuitously identified PAM, another group diagnosed by family investigations and a third group representing a more severe disease.
CT findings in children are less typical compared to adults and show more diffuse lung involvement although patchy involvement has been also observed. Typical features are ground-glass opacities, septal thickening and bilateral micronodular densities and calcifications mostly present along the bronchovascular bundles, the interlobular septa and the subpleural area. Calcifications are frequent, but contrary to adults, are less severe and smaller and mainly restricted to the lower lobes. Ground-glass opacities that are rarely seen in adults were more common in the children younger than 6 years of age, and children never show signs of lung fibrosis [7].
Diagnosis
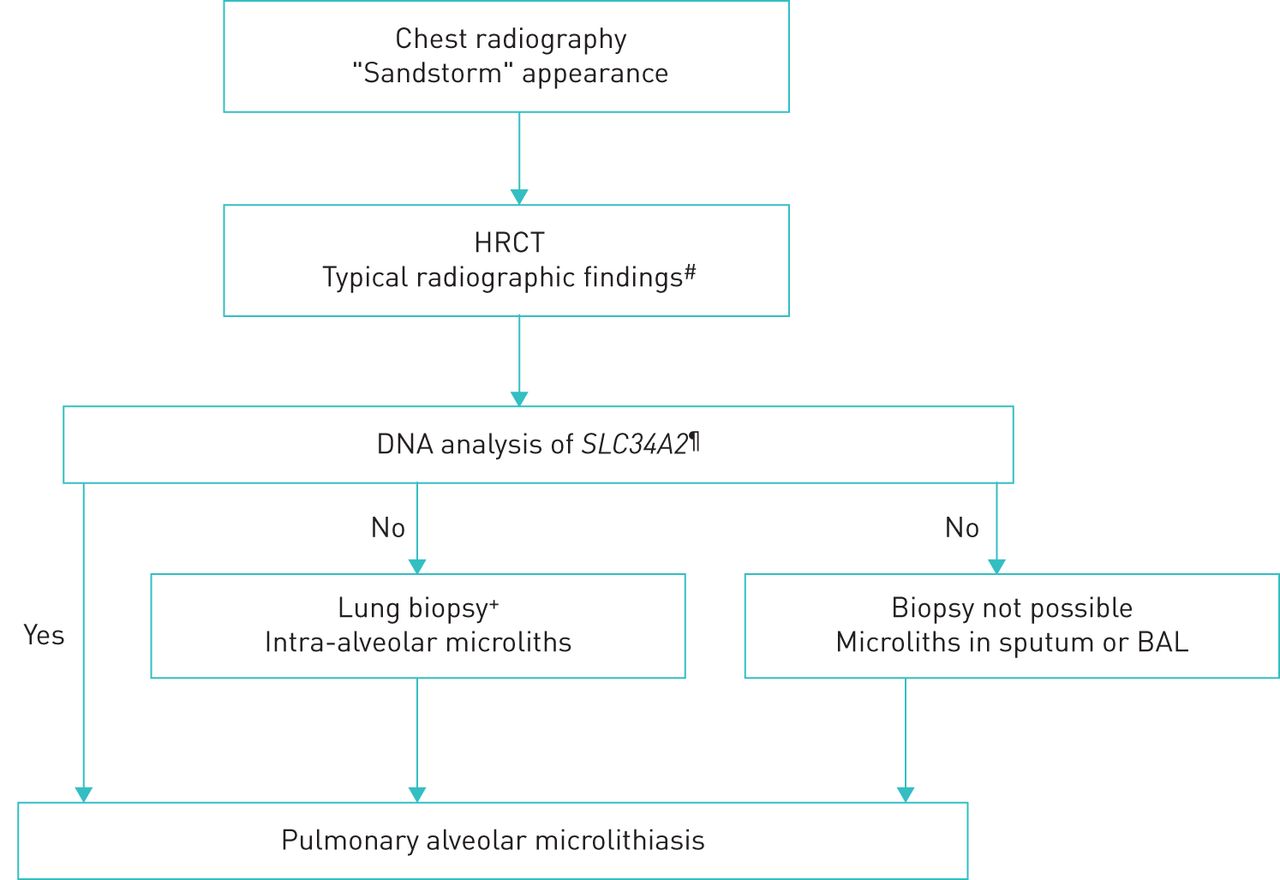
A diagnosis of PAM can confidently be based on typical radiographical findings and genetic testing proving rare biallelic SCL34A2 gene variants. When genetic testing is unavailable or negative, a definite diagnosis can be established by bronchoalveolar lavage (BAL) or a lung biopsy, either bronchoscopic or surgical, with the demonstration of intra-alveolar microliths (figure 1). The decision to obtain tissue should preferably be taken at a multidisciplinary team discussion at experienced centres and individualised based on patients' individual risk and availability and experience of procedures at the centre. Transbronchial forceps biopsies generally results in smaller biopsies with crush artefacts whereas transbronchial cryobiopsies and surgical lung biopsies are larger with a higher diagnostic yield but also with a risk of more complications [49, 50].
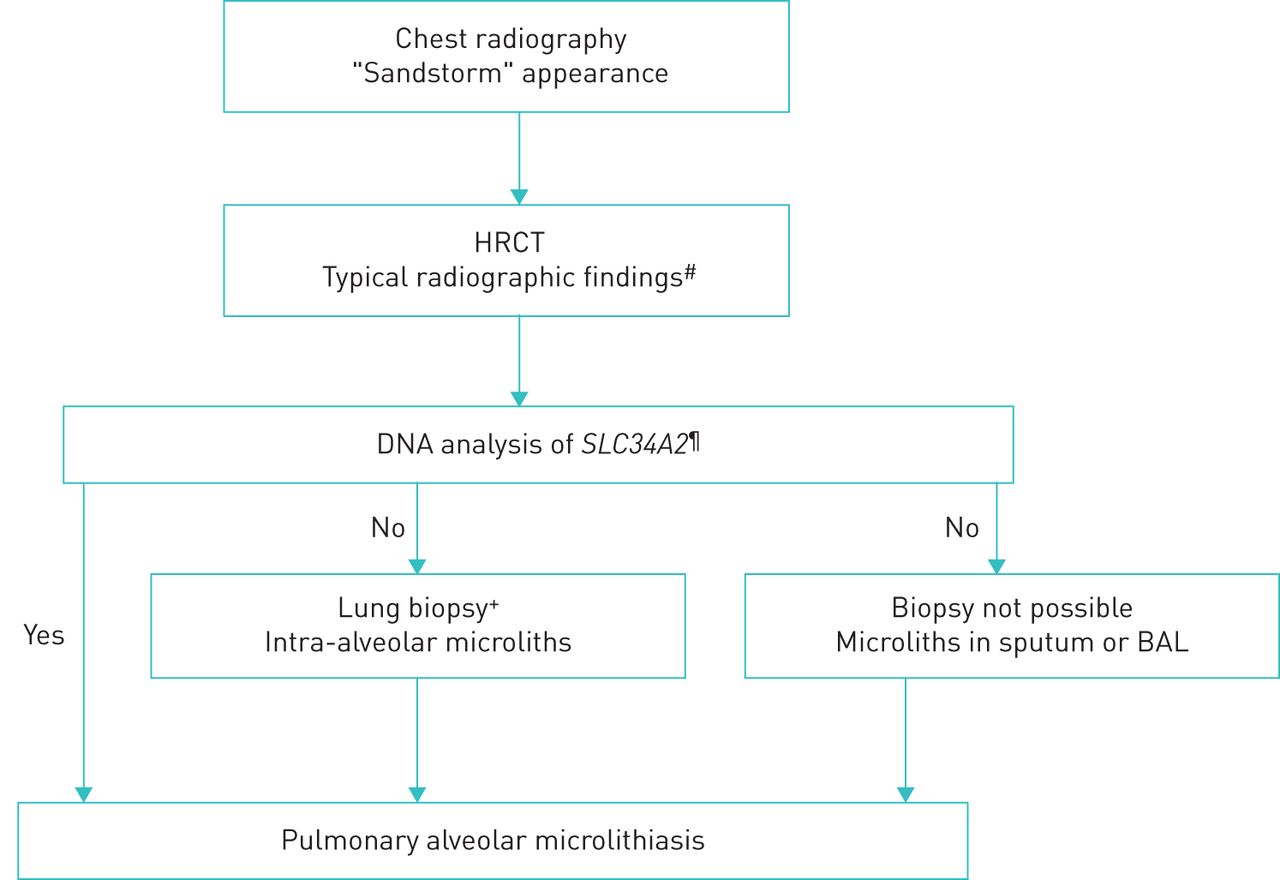
Diagnostic algorithm for pulmonary alveolar microlithiasis. The algorithm is step-wise with the least invasive procedure preferred as the first choice. The decision to obtain tissue should always be individualised and preferably be taken at a multidisciplinary team discussion. Less experienced centres are recommended to consult an expert centre. #: intra-alveolar calcifications, micronodules, ground-glass opacities, thickened septa, subpleural cysts; ¶: if genetic analysis is not available, proceed to invasive examinations;+: transbronchial forceps or cryobiopsy; the choice between these should be based on availability and experience and considering the least invasive procedure first; if transbronchial biopsies are non-diagnostic, consider surgical lung biopsy.
Blood assays
Standard blood tests have no place in PAM diagnostics, as common biomarkers of bone metabolism in peripheral blood including, but not limited to calcium and phosphate, are normally not affected in PAM [18, 40]. Blood tests for genetic analysis showing a variant in SCL34A2 in homozygous state or two variants in compound heterozygous state will provide the diagnosis.
Surfactant protein (SP)-D, SP-A and monocyte chemotactic protein 1 (MCP-1) have all been suggested as useful serum biomarkers of disease activity but are not routinely used in most centres [17, 51, 52].
Sputum testing
Sputum examination for microliths has shown presence of microliths in older studies but is rarely used nowadays [53]. More contemporary, DNA extracted from sputum can be used for genetic analysis.
Lung function
Pulmonary function tests will most often present as normal in less advanced disease but as PAM progress a restrictive lung function impairment with reduced dynamic and static volumes and diffusion capacity will develop [18, 40].
Imaging
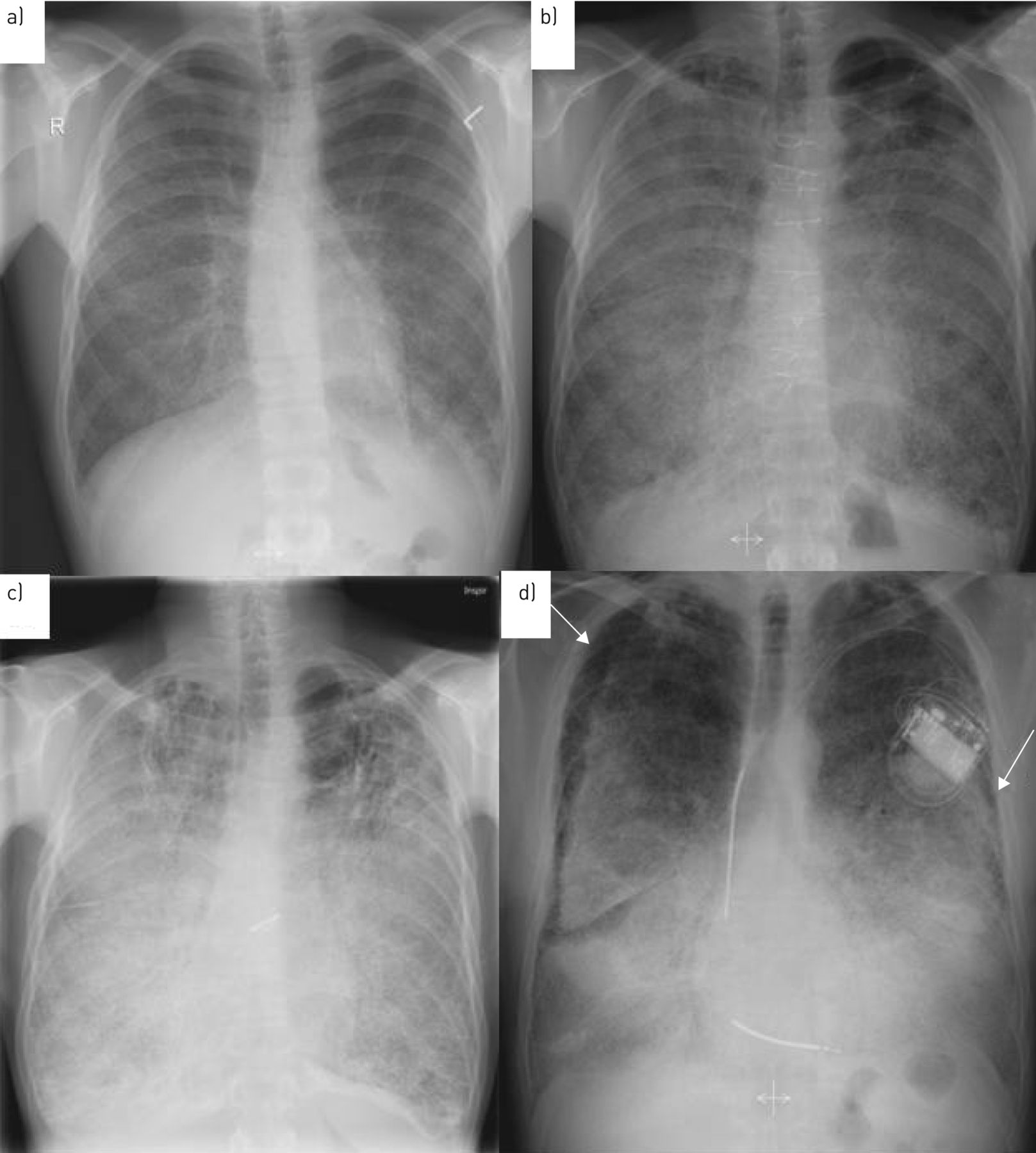
On chest radiography, PAM is characterised by a “sandstorm” appearance with diffuse distribution of innumerable small sand-like micronodules (also called microliths, calcipherites or calcospherites), within the air spaces, particularly in the lower parts of the lung. A black pleural sign or black pleural line is a common finding and describes the vertical strip of peripheral hyperlucency between the ribs and the adjacent diffusely dense calcified lung parenchyma and is caused by subpleural cystic changes. Often, the mediastinal contour and diaphragm cannot be identified due to intensive microliths and interstitial involvement in advanced disease [54] (figure 2).

Chest radiography of two patients with pulmonary alveolar microlithiasis. a) Patient 1, 2003, radiological severity stage II. b) Patient 1, 2013, radiological severity stage III. c) Patient 1, 2019, radiological severity stage IV. d) Patient 2, 2007, radiological severity stage IV. White arrows demonstrate the “black pleural sign”.
High-resolution CT is regarded as the gold standard for PAM diagnosis and demonstrates numerous sandstorm-like calcifications throughout the lungs with subpleural and peribronchial distribution particularly affecting the basal segments and paracardiac region [54]. Typical features include intra-alveolar calcifications (microliths), micronodules, ground-glass opacities along with superimposed smooth thickened of the interlobular septa, subpleural microhoneycombing cystic changes, subpleural reticular thickening and, in some instances, a crazy paving pattern. Calcifications of the pleura also have been reported [54, 55] (figure 3).
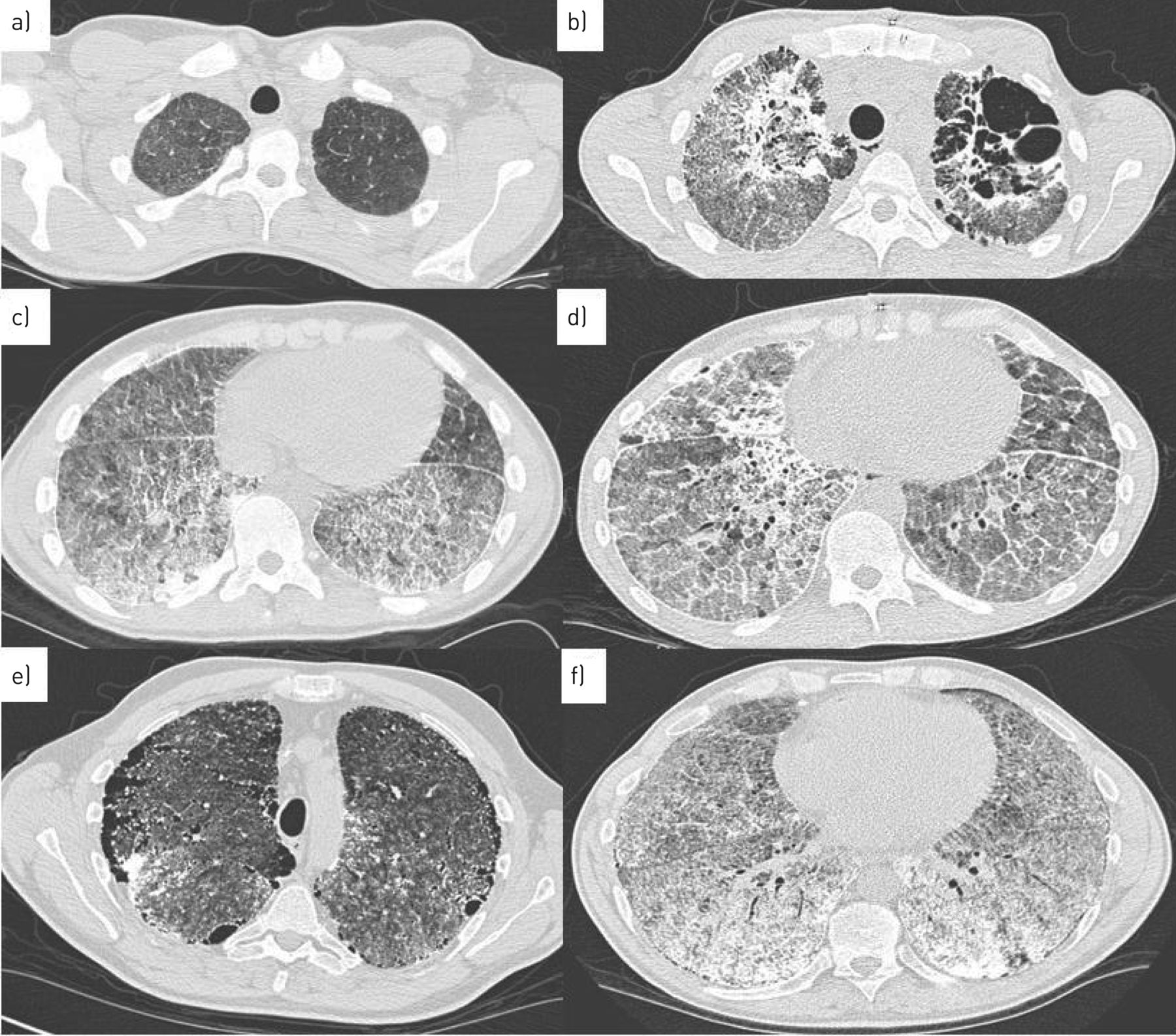
High-resolution computed tomography of two patients with pulmonary alveolar microlithiasis. a) Patient 1, 2003, ground-glass opacities in apical lung fields and c) calcifications, micronodules, thickened septa in basal lung fields. b) Patient 1, 2016, calcifications with micronodules, thickened septa and cysts in apical lung fields and d) progression of calcifications with consolidations and crazy paving. e) Patient 2, 2005, radiological severity stage IV. Subpleural microcysts (black pleural sign), ground-glass opacities and micronodules in apical lung fields and f) same patient, basal lung fields showing extensive calcifications with thickened septa and micronodules.
Recently, four radiological severity stages have been proposed:
Stage 1: Microliths are few and the calcification grade is low. The stage is therefore known as the “pre-calcific phase” and is only rarely observed in asymptomatic children.
Stage 2: The typical “sandstorm” appearance is now present. The calcific micronodules (<1 mm in diameter) are seen scattered throughout the lungs with basal and mid-lung predominance. Some micronodules may be larger in diameter (∼2–4 mm). The boundaries of the heart and diaphragm appear clear. Stage 2 is characteristic in childhood or adolescence.
Stage 3: In the third stage the micronodules disseminate, interstitial thickening is observed and the heart and diaphragm boundaries are obliterated. This phase is often seen in young adults.
Stage 4: This is the most advanced stage and is characterised by extensive interstitial calcification and sometimes involvement of the pleura giving the appearance of “white lungs”. Fibrosis, subpleural microcysts, areas of ossification and pneumothorax can be present. This stage is typically reported in older patients or in cases with advanced disease. This appearance is very characteristic and can in the right clinical context be sufficient for diagnosis [6].
Thoracic ultrasonography has gained increasing interest in recent years and increased thickening and irregularities of a hyper-echoic pleural line and millimetric echogenic foci with no marked acoustic shadowing in the subpleural and basal fields area has been described [41]. However, discrepancy of thoracic ultrasonography findings compared with conventional CT imaging limits its use so far [41, 56].
Histological features
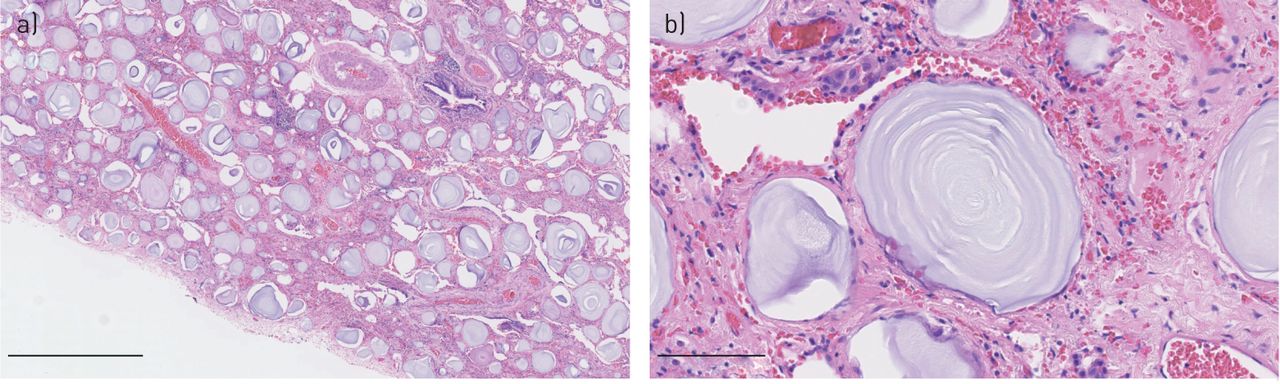
Typical histopathological findings are best seen with trichrome staining and characterised by multiple intra-alveolar spherical calcium and phosphate deposits, the so-called calcospherites or microliths (figure 4). Microliths are composed of hydroxyapatite crystals that form concentric lamellae surrounding an amorphous central nucleus. The size is usually less than 1 mm, but occasionally larger microliths up to 5 mm are observed. Inflammation, fibrosis and calcification of the lung interstitium and pleura are also noticed. The histopathological findings of intra-alveolar microliths are most often very characteristic. In cases of less classical findings, consultation with an expert centre is recommended.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Lung section from a male patient with pulmonary alveolar microlithiasis showing calcified microliths in the intra-alveolar spaces. Haematoxylin and eosin staining. Original magnification: a) ×50 and b) ×400. Scale bars: a) 1 mm and b) 100 μm. By courtesy of pathologist Johanne Lade Keller, Dept of Pathology, Aarhus University Hospital, Aarhus, Denmark.
Analysis of BAL is often inconclusive but occasionally, mild lymphocytosis and microliths can be identified in BAL; microliths can occasionally be found in sputum samples [18, 34, 40, 57].
PAM clinical disease severity score
A clinical disease severity score has recently been proposed. The score is based upon a composite assessment of nine clinical parameters: pulmonary hypertension, dyspnoea, chest pain, fatigue, clinical progression, limitations of daily activities, incapability to work, forced vital capacity and diffusing capacity of the lung for carbon monoxide, and determines three severity grades: mild, moderate and severe [18]. The score needs still validation in other and prospective cohorts.
Differential diagnosis
On imaging, the differential diagnosis includes a number of diseases with a similar radiological appearance of micronodules, such as miliary tuberculosis, fungal infection, varicella pneumonia, sarcoidosis, amyloidosis, pulmonary alveolar proteinosis, pulmonary hemosiderosis, metastatic calcification in chronic renal failure and pneumoconiosis (e.g. silicosis, talcosis, stannosis, baritosis) [6, 58–60]. It is important to differentiate PAM from these diseases to ensure accurate treatment. However, while the radiological appearance of calcifications is more intense in patients with PAM, the symptoms of the other differential diagnoses are usually more severe.
Treatment
Currently, there is no approved medical or genetic treatment available to alter the progression of PAM and many therapies, including corticosteroids, calcium-binding agents and bronchoalveolar whole-lung lavage, have proven to be ineffective. The long-term prognosis is poor in some patients and lung transplantation is the only effective therapy to date.
Corticosteroids
Corticosteroids have been reported to result in symptom alleviation in a few patients with PAM but are in general considered noneffective [46, 61–63].
Sodium thiosulfate
Sodium thiosulfate is a calcium-chelating and solubilising agent that has been tried in a single patient with PAM. Despite treatment, disease progression was observed on chest CT [64].
Whole-lung lavage
As the microliths are located in the alveolus, repeated BAL has been suggested as a possible treatment strategy for PAM but it has so far been ineffective [39, 61, 65].
Low-phosphate diet
A low-phosphate diet in mice with a PAM phenotype has shown reduced formation of microliths but there are so far no human studies and a potential beneficial effect is thus unknown in PAM patients [17].
Bisphosphonates
Bisphosphonates inhibit bone resorption by attaching to hydroxyapatite binding sites in the bone and by interfering with the osteoclasts [66]. It has been speculated whether bisphosphonates could be beneficial in patients with PAM by preventing calcifications in the alveoli and hence the formation of microliths [66–68]. A total of 12 cases of patients with PAM treated with bisphosphonates have been published [23, 40, 42, 67–74]. The results are inconsistent as some patients responded with respect to clinical and radiological improvement, while others remained unaffected. Most patients (10) were treated with etidronate with a greatly varied dose (200 to 900 mg·day−1) and treatment duration (6 months to 11 years). Insufficient dosage, treatment duration and initiation of treatment in patients with advanced disease with well-formed microliths have been considered as possible explanations for treatment failure.
Supportive treatment
Patients with PAM who are hypoxemic should be prescribed supplemental oxygen therapy. Pneumococcal and influenza vaccinations should be offered to all PAM patients.
Lung transplantation
Lung transplantation, either single or bilateral, is usually considered in patients with end-stage lung disease. The optimal time for referring patients with PAM for lung transplant has not been defined but referral is recommended before the onset of severe right ventricular dysfunction [6].
So far, lung transplantation has been reported about 20 patients. Recently, Alrossais et al. [75] reviewed 19 lung transplant patients with PAM. The mean age at transplantation was 47 years, with an average follow-up period of almost 3 years. Complications and survival seemed not to differ from those described in transplant recipients with other background lung diseases. Mortality was caused by bronchiolitis obliterans (n=2), sepsis (n=2), haemodynamic instability (n=1), and multi-organ failure (n=1) [75].
The longest survival continued follow-up beyond 15 years [76]. No recurrence of PAM has been observed during the post-transplant period.
Familial genetic counselling
Families with PAM should be offered genetic counselling to help them understand the implications of having a genetic disease. In addition, prenatal testing and pre-implantation genetic diagnosis for PAM may be offered to families at high risk for having a child with PAM.
Prognosis
The disease course of PAM is variable and there is lack of observations to inform the long-term prognosis [18]. However, case series suggest that the disease is slowly progressive with reduced survival. Some patients with PAM can remain stable and the longest reported follow-up period is 58 years [6]. If a diagnosis was made in childhood, there are a few reports with more than 40 years of follow-up [6]. A Japanese study of 53 patients with PAM found that 18 patients (34%) died due to PAM within a follow-up period of 10–49 years. The mean age at death was 46 years [77]. Similarly, others also report death to occur in the fifth decade of life, resulting from respiratory and right heart failure [6].
Environmental factors like smoking, inhalation of snuff, repetitive lung infections and cold weather may be associated with both the onset and the course of the disease but firm evidence is lacking and further investigations are warranted [19, 27, 35, 40, 76].
Future perspectives
PAM remains a challenge due to its rareness and limited treatment options. In recent years, our knowledge of clinical characteristics, risk factors, genetics, pathogenesis and diagnostic approaches has increased, but major gaps are still present. Still, it is case reports, case series and retrospective reviews that form our knowledge. Larger patient cohorts based on international collaborations as in the recent published paper by Jönsson et al. [18] or international patient registries are needed to collect cases, allow for biobanking and for execution of prospective studies. Characterisation of pathogenic mechanisms and possible genotype–phenotype correlations is essential to explore the relevance of different genetic variants in patients as is also identification of risk factors for disease progression and extrapulmonary involvement. Increased awareness and better access to genetic testing is warranted to avoid lung biopsies as these are inevitably associated to risk of complications. Biomarkers and predictive tools for disease progression and prognosis are needed to inform patients of their disease course and to decide when to initiate potential future effective treatment and to refer patients for lung transplant evaluation. Also, a disease severity score will be useful for evaluation of disease burden, stratification of patients and for research design. Ultimately, we need better treatment options. Increased knowledge of the functional deficits of different variants and discovery of the crystalline structure of NaPi-2b will be steps on this pathway.
Conclusion
PAM remains a rare disease with fewer than 1100 cases reported. It is an autosomal recessive disease and 30 allelic variants have been reported in around 60 cases. The types of variants are heterogeneous and located almost in all exons of the SLC34A2 gene. The disease spectrum is variable and although a PAM disease severity score has been proposed, a validated genotype–phenotype correlation has to be proven. Until now, the treatment options are few and lung transplantation remains the only effective therapy. Increased knowledge of the genetic background will result in increased understanding of the pathophysiology of PAM and will hopefully result in new treatment options in the future.
Footnotes
Conflict of interest: E. Bendstrup has nothing to disclose.
Conflict of interest: Å.L.M. Jönsson has nothing to disclose.
- Received May 20, 2020.
- Accepted June 16, 2020.
- Copyright ©ERS 2020
This article is open access and distributed under the terms of the Creative Commons Attribution Non-Commercial Licence 4.0.
References










