Article Text

Abstract
Background: Neuro-Sweet disease is a rare condition of central nervous involvement accompanied by cutaneous Sweet lesions. Neuropathological changes in neuro-Sweet disease are unknown.
Objective: To describe post-mortem findings of the first case of neuro-Sweet disease.
Results: A 44-year-old Japanese man developed recurrent episodes of cerebral and brainstem encephalitis with cutaneous Sweet lesions from the age of 34 years. His HLA typing was B54 and Cw1, and the symptoms and MRI abnormalities markedly subsided following corticosteroid therapy. Histologically, there were multiple lesions of perivascular cuffing of small venules by macrophages without vasculitis in the thalamus, temporal lobe, basal ganglia, pons, leptomeninges or ventricular ependym.
Conclusions: The core neuropathological findings were: perivascular cuffing around particularly small veins; absence of granulomatous or necrotic angitis; mainly macrophage infiltration; and the thalamus being most affected. In the present case, the diagnosis of neuro-Sweet disease was made by skin biopsy 5 years after the onset of the central neuron system symptoms. We should pay more attention to skin lesions in steroid responsive recurrent encephalitis in patients who are HLA-B54 or Cw1 positive.
Statistics from Altmetric.com
In 1964, Sweet described eight patients with four cardinal features, including fever, peripheral neutrophil leucocytosis, raised painful plaques and a dense dermal infiltration with mature neutrophils.1 This peculiar “febrile neutrophilic dermatosis” of unknown aetiology, which partly resembles Behçet disease, is currently called Sweet disease. The concurrence of malignancy (haematological neoplasia and various cancers) or systemic inflammatory diseases (inflammatory bowel disease and rheumatoid arthritis) was reported in Sweet disease.2 Also, many medicines (granulocyte colony stimulating factor, proteasome inhibitor bortezomib, carbamazepine, etc) were reported as causes of drug induced Sweet syndrome.3 Although it rarely affects the central nervous system, Hisanaga et al4,5 reviewed cases of Sweet disease with central nervous system involvement, and proposed a concept, “neuro-Sweet disease”, that is distinct from neuro-Behçet disease. This paper reports the first autopsy case of neuro-Sweet disease.
CASE REPORT
A 34-year-old man developed high fever and general fatigue in July 1990. Double vision, dysarthria, right-sided slight hemiparesis and hemihyperesthesia, and disturbance of consciousness appeared after several days. His symptoms progressed subacutely and brain CT on the fifth day after onset revealed multiple low intensity lesions in the brainstem, basal ganglia and cerebral white matter. His general physical examination on the 11th day after onset was unremarkable, and body temperature was 36.2°C. Neurological examination showed mild disturbance of consciousness, truncal ataxia, left blepharoptosis, double vision with limitation of left eye adduction, dysarthria and right hemiparesis. The deep tendon reflex was hyperactive and the Babinski sign was positive on the right. He showed no meningeal signs. Haematological examination showed that the white blood cell count was 10.41×109/l, with 6.14×109/l neutrophils. Erythrocyte sedimentation rate, C reactive protein level and serum chemistry data were within normal limits. Several autoantibodies were negative. Cryoglobulin and angiotensin 1 converting enzyme were within normal limits. Culture of blood samples for bacteria and fungi, and antibodies against several viruses were undetectable in serum samples. A lumbar puncture disclosed moderate pleocytosis (87 cells/mm3, 94% lymphocytes and 6% neutrophils without malignant cells). Protein content was 51 mg/dl, and glucose level was 63 mg/dl.
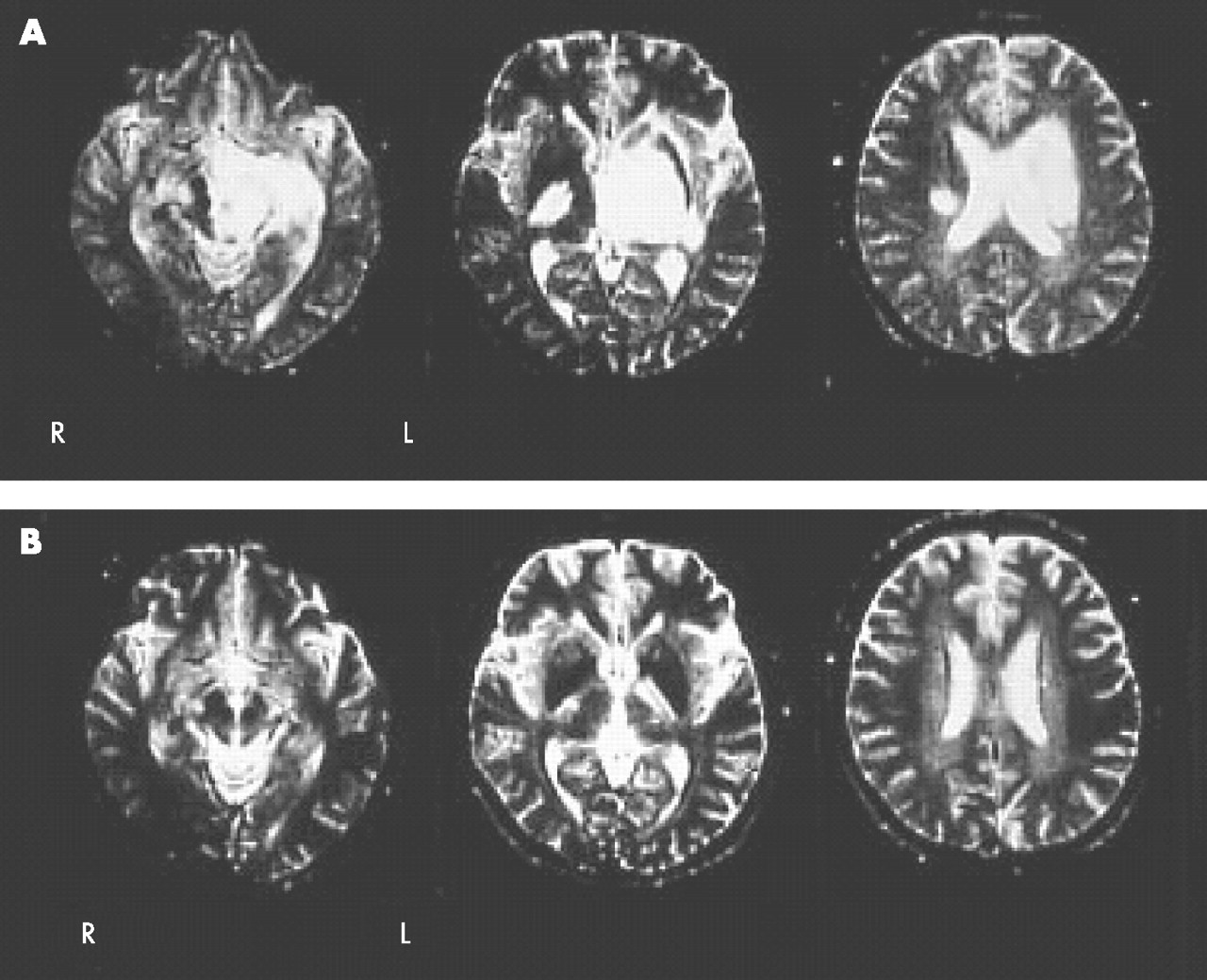
MRI of the brain on the 12th day after onset revealed multiple large and small lesions with high signal intensity on T2 weighted images and normal or low signal intensity on T1 weighted images in the brainstem, thalamus, basal ganglia and cerebral white matter without gadolinium enhancement (fig 1A). Electroencephalography showed basic activity of 6–7 Hz theta waves with abundant 4–5 Hz slow theta and delta activities, with a left-sided predominance. A diagnosis of encephalitis of parainfectious nature was made, and he was treated with antibiotics, acyclovir (750 mg/day) and betamethasone (12 mg/day). His symptoms and MRI findings (fig 1B) improved markedly in 2 weeks. Similar but milder encephalitis recurred six times between January 1993 and September 1999. The clinical symptoms and abnormal findings on MRI dramatically responded to corticosteroids, and he recovered almost completely each time.

Brain MRI at the first admission. (A) MRI taken on the 12th day after onset of the disease shows abnormal high signals in the midbrain, pons, thalamus, basal ganglia, temporal lobe, internal capsule and cerebral white matter. The left half is more severely affected throughout the brain. (B) MRI taken after corticosteroid therapy on the 29th day after onset of the disease shows almost complete disappearance of abnormal high signals.
Erythema nodosum-like painful eruptions appeared for the first time in October 1995 (fig 2A, B), and skin biopsy was performed in September 1999 when erythema nodosum-like painful eruptions recurred. Histopathologically, the epidermis was spared, and oedema of the papillary dermis was seen. Moderate infiltrations of neutrophils and monocytes surrounded the small blood vessels in the superficial and mid-dermis. There was no necrotising vasculitis (fig 2C, D). These findings were consistent with the skin plaques of Sweet disease. On July 2000, he was found dead.

Erythematous eruptions. (A, B) Erythematous eruptions on his face and extremities in September 1999. (C, D) Histological pictures of the erythematous eruption showing dense infiltration of neutrophils in the dermis without vasculitis. (C: bar = 400 μm, D: bar = 100 μm, haematoxylin–eosin stain). Informed consent was obtained for publication of this figure.
During his clinical course, erythema nodosum-like painful eruptions appeared temporally on the legs, back, hands and face. Oral aphthae temporally appeared in July 1997 and September 1999, and a pathergy test was elicited in September 1999. Uveitis and genital ulceration were absent throughout his clinical course. His HLA typing was A24, A21, B44, B54 and Cw1.
PATHOLOGICAL AND NEUROPATHOLOGICAL FINDINGS
Autopsy was performed approximately 15 h after death. General pathological examination revealed acute lung congestion and cardiomegaly indicating sudden circulatory failure. Any pathological findings that would have caused sudden death were not found in the heart, lung or other organs.
Neuropathological findings
Macroscopic findings
The formalin fixed brain weighed 1330 g. The brain was swollen moderately. The leptomeninges were opaque and thickened with severe fibrosis, and adherent to the brain surface, particularly in the basal portion of the brain. The coronal sections showed many congested veins associated with very small haemorrhages in the cerebral white matter, basal ganglia, thalamus, pons, medulla, cerebellar white matter and dentate nucleus. The brainstem was mildly atrophic. There were no focal abnormal lesions or findings of cerebellar tonsillar or uncal herniation.
Microscopic findings
Klüver–Barrera stain revealed areas of pallor and foci of various sizes in the cerebrum and brainstem, which were indistinctly demarcated from the normal areas and disseminated asymmetrically. In high power observations, the pallor areas consisted of loss of myelin frequently associated with microscopic infarction and haemorrhages around the small vessels. On Bodian stain, axons were relatively preserved in the cerebral white matter that was pale on Klüver–Barrera stain. Loss of myelin in the white matter was considered to be due to tissue breakdown rather than primarily demyelination. Haematoxylin–eosin stain showed perivascular cuffing and infiltration into the surrounding areas forming nodules. These perivascular cuffings were most marked in the thalamus (fig 3A) although the temporal lobe, basal ganglia, pons and leptomeninges (fig 3B) were similarly affected. Both the white matter and cortex were primarily affected. Neurons of the cortex and thalamus were relatively preserved, but there were a few neuronophagia and mild neuronal loss. The vessel walls appeared almost normal, and features indicating granulomatous or necrotising angitis were absent. Elastica van Gieson stain (fig 3C) clearly revealed that perivascular cuffing affected small veins more severely than small arteries. Leucocyte immunohistochemistry was performed using anti-CD3 antibody for T cell, anti-CD20 antibody for B cell, HLA-DR for microglia and anti-CD68 antibody for macrophages. It revealed that infiltrating cells consisted of mainly macrophages (fig 3D).

{kind=link}
{kind=link}
{kind=link}
Histopathological and immunohistopathological findings. (A) Perivascular cuffing and monocyte infiltration into the substrate around the vessels are seen in the thalamus (bar = 300 μm, haematoxylin–eosin stain). (B) Many inflammatory cells surround small vessels, more markedly veins, and infiltrate into the subarachnoid spaces of the leptomeninges of the right medial temporal lobe (bar = 200 μm, haematoxylin–eosin stain). (C) Elastica van Gieson preparation of the basal ganglia. Veins are more extensively affected by inflammatory cells than arteries (bar = 300 μm). (D) Inflammatory cells mainly consist of macrophages. The basal ganglia (bar = 200 μm, CD68 immunostain).
DISCUSSION
There are many overlaps in the clinical symptoms of Sweet disease and Behçet disease. Oral aphthae, genital ulcers, erythema nodosum-like eruptions, pathergy test and uveitis are encountered in both diseases. Distinct categorisation has only been achieved on the basis of the histopathology of skin lesion. HLA-Bw54 and Cw1 are significantly higher in patients with Sweet disease,6 and HLA-Bw51 in patients with Behçet disease.
Recently, Hisanaga et al reviewed the clinical findings of 42 cases of neuro-Sweet disease and proposed diagnostic criteria on the basis of neurological, dermatological, other features and HLA association.5 According to these authors, benign clinical course, good responsiveness to systemic corticosteroid therapy, absence of vasculitis in the skin lesions and HLA-Bw54 and/or Cw1 are important for a diagnosis of neuro-Sweet disease.
Our patient fulfilled all of the essential features of neuro-Sweet disease, and clinically probably had neuro-Sweet disease. In addition, the clinical features of this patient did not meet the criteria for a diagnosis of Behçet disease.7 Although vasculitis can occur in skin lesions in Sweet disease,8 erythema without vasculitis seen in this case was differentiated from the skin lesions seen in Behçet disease accompanied by vasculitis.9 The skin lesions in this case had monocytes in addition to neutrophils which are not recognised in classical descriptions of Sweet disease. However, recent publications have demonstrated the presence of immature mononuclear granulocytes in the skin lesions of Sweet disease.10
The core neuropathological findings in our patient were as follows: (1) perivascular cuffing around particularly small veins, frequently associated with microscopic haemorrhage, infarctions and loss of myelin; (2) absence of granulomatous or necrotic angitis; (3) mainly macrophage infiltration; (4) the thalamus was affected most; and (5) there were a few neuronophagia and mild neuronal loss in the cortex and thalamus.
In neuro-Behçet disease, inflammation is widespread but more marked in the brainstem. Parenchymal injury is obvious, and neuronal necrosis and atrophy of the cerebellum, brainstem and spinal cord are occasionally present. These changes indicate that neuro-Behçet disease is refractory to immunosuppressive therapy. Necrotic vasculitis is not an essential finding in neuro-Behçet disease. The parenchymal and perivascular lymphocytic infiltrates are exclusively T cells.11 In the early phase of encephalitis, inflammation extends from the pial surface through the cerebral cortex and into the white matter. Necrotic cells, foci of haemorrhage and an intense perivascular and interstitial infiltrate of lymphocytes and macrophages are seen.12 Active plaques of multiple sclerosis are hypercellular lesions containing a relatively dense perivascular and parenchymal infiltrate of lymphocytes and macrophages, and scattered active astrocytes. The inflammation tends to be greatest towards the edge of the plaque and in the contiguous intact white matter. The lymphocytes in these regions are mostly T cells.13 Destruction of the brain substrates in the present case was minimal, probably corresponding to the dramatic response to corticosteroid therapy.
Acknowledgments
The wife of the patient consented to the publication of the pictures of the skin lesions. The authors thank the following colleagues for help during the clinical data collection: M Matsuyama, MD, K Suzuki, MD, T Ohira, MD, K Matsumoto, MD, S Nishida, MD, K Yada, MD. We also thank Ms Hisami Akatsuka for her technical assistance in tissue preparations for histopathology.
REFERENCES
Footnotes
-
Funding: This study was partly supported by a Grant-in-Aid from the Research Committee of CNS Degenerative Diseases, the Ministry of Health, Labor and Welfare, Japan and by a Grant-in-Aid for the Scientific Research from the Ministry of Education, Science, Sports and Culture, Japan.
-
Competing interests: None.
-
Informed consent was obtained for publication of fig 2.