Article Text

Summary
A male neonate with maternally inherited Marfan syndrome was also diagnosed with Down syndrome at 3 weeks of age. To our knowledge this is the first described case in the literature of the co-occurrence of Down syndrome and Marfan syndrome in a neonate. The diagnosis of Down syndrome was delayed and we hypothesise that Marfan syndrome had masked the usual phenotypic features of Down syndrome. The phenotype of this child is intriguing and has lead to speculation of the possible interaction of the two syndromes.
Statistics from Altmetric.com
Background
The co-occurrence of Marfan syndrome (MFS) and Down syndrome (DS) has not been previously identified in a neonate. We describe the features and clinical course of a baby whose mother had MFS and genetic testing after birth revealed not only inheritance of the maternal fibrillin 1 gene (FBN1) mutation but also trisomy 21. Apart from hypotonia, the patient had no typical features of DS and we postulate that the phenotypic expression of DS has been masked by the co-occurrence of MFS. Furthermore, there is overlap of the phenotypes in the two syndromes, as congenital cardiac and diaphragmatic abnormalities described in this case could be attributable to either MFS or DS.
Case presentation
A male infant weighing 3.1 kg was delivered at 42 weeks gestation by emergency caesarean section for a poor cardiotocography trace. There was a history of maternal MFS, with the mother being diagnosed at 32 years of age with the condition. She was not severely affected though had had surgery at age 8 for scoliosis. There were no other antenatal concerns and the antenatal anomaly scans were normal. Parents opted for the fetus not to be tested for MFS antenatally and they were reassured that the fetus should not be phenotypically more affected than the mother. There were no maternal risk factors for sepsis.
At birth he was noted to have poor respiratory effort with central cyanosis requiring resuscitation and intubation and was transferred to paediatric intensive care unit (PICU). Over the next few hours he needed ventilatory support and had high oxygen requirements. An echocardiogram (ECHO) showed he had persistent pulmonary hypertension in the setting of a dysplastic tricuspid valve with severe tricuspid regurgitation. He also had biventricular hypertrophy and a patent foramen ovale (PFO). During the next few weeks he remained intubated and ventilated with significant oxygen requirements until day 12 of life. He was treated with nitric oxide and had inotropic requirements. He was also started on diuretics for cardiac failure and developed acute renal failure requiring peritoneal dialysis. Empirical antimicrobial therapy was also started but stopped at 5 days when there was no microbiological evidence for sepsis. He was weaned from his inotropic and renal support by day 13 of life and began to make a steady recovery.
During his stay in PICU he was noted to be hypotonic with minimal spontaneous movements. This was initially thought to be secondary to sedation and paralysis; however, the hypotonia persisted even during his recovery phase. He also had the following dysmorphic features: large hands and feet with arachnodactyly, wide carrying angle of elbows, ulnar deviation of wrists and small simple ears. In view of the hypotonia and dysmorphic features, chromosomal and mitochondrial studies were performed, which not only confirmed a FBN1 mutation but indicated that he had trisomy 21.
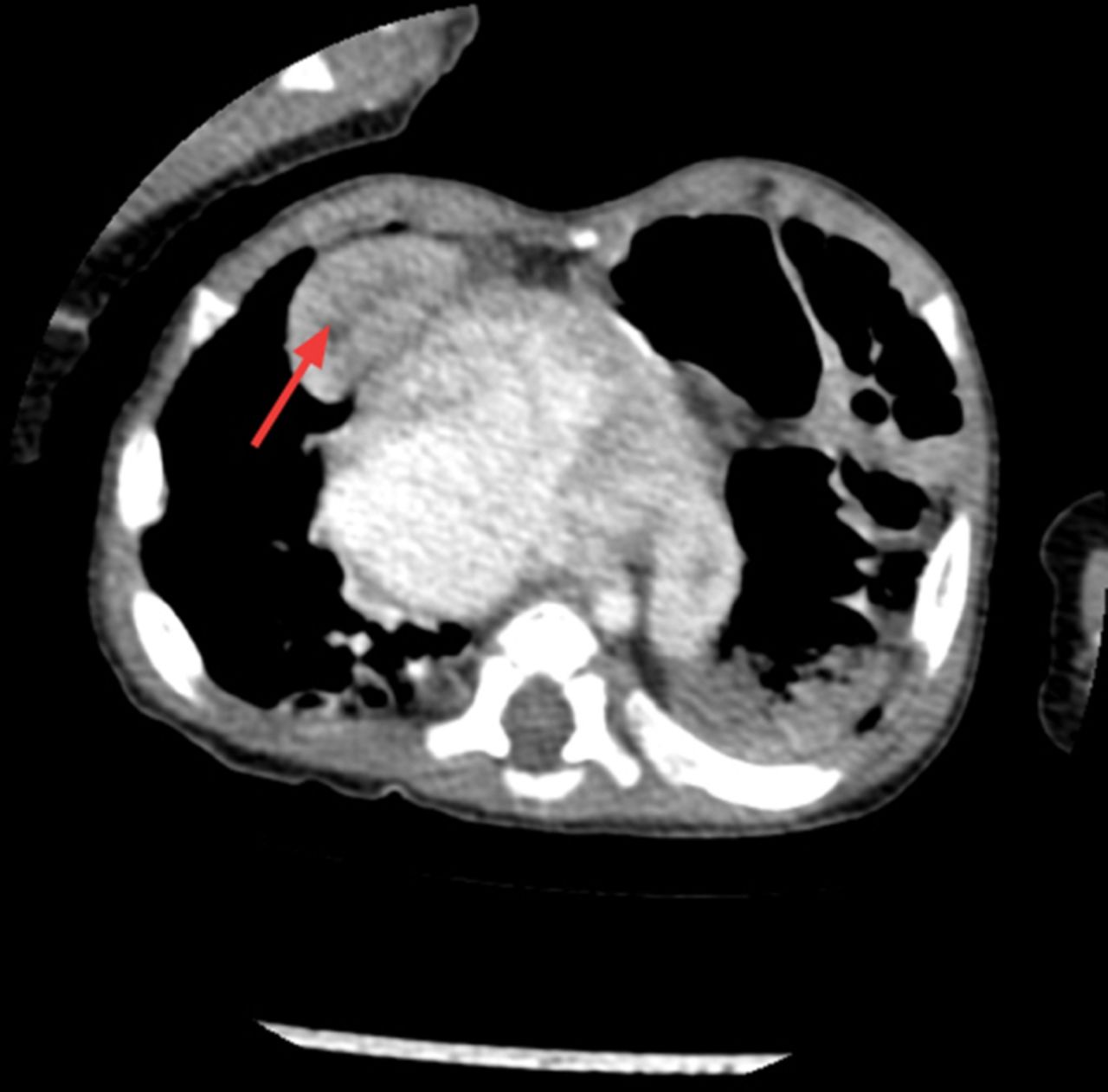
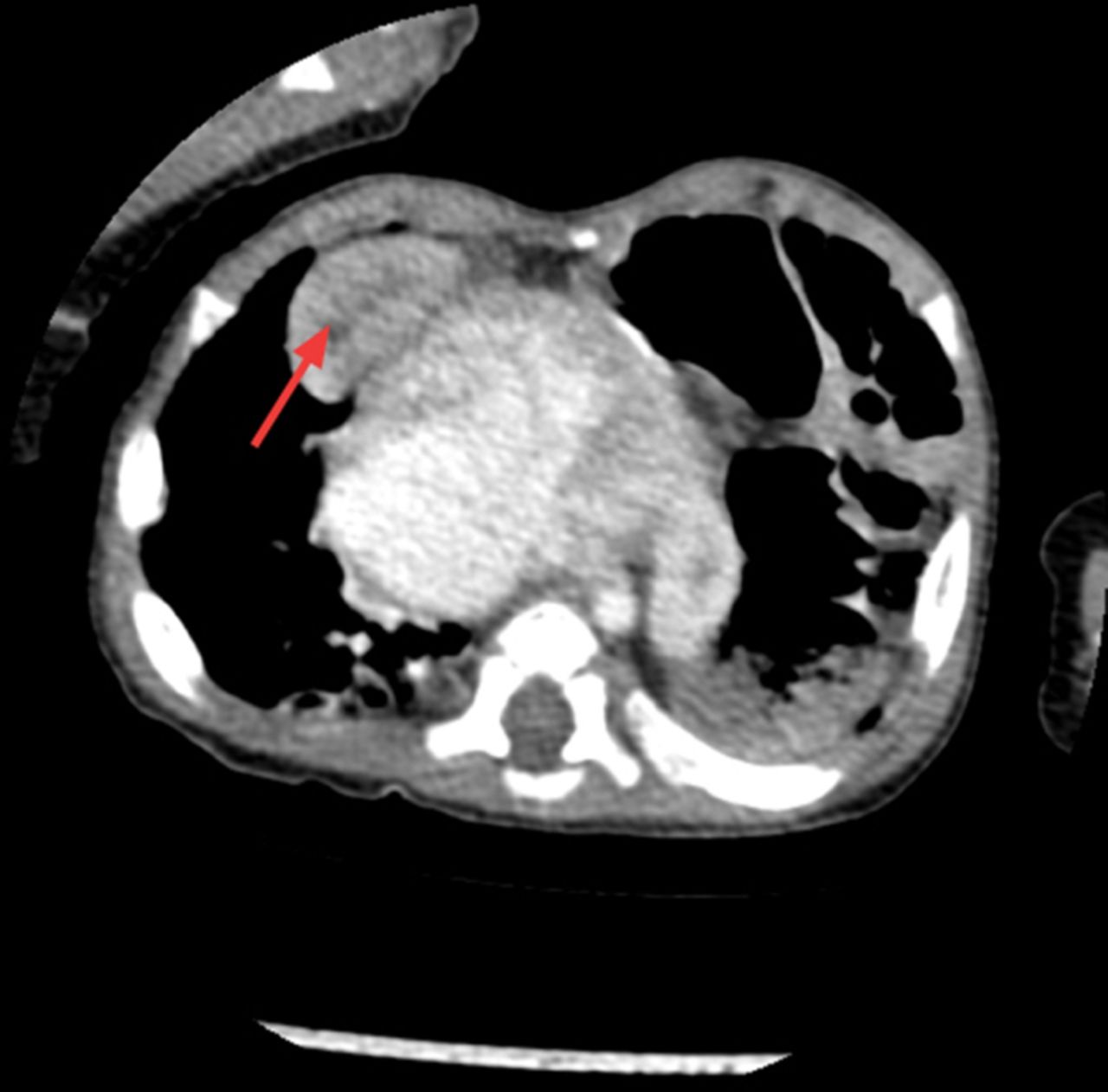
He was discharged home on day 26 of life being fully breast fed, self ventilating in air and on no medications. He was followed up at regular intervals by the paediatric cardiology team. A repeat ECHO post discharge at 6 weeks of age showed closure of PFO but persistently elevated pulmonary pressures and moderate tricuspid regurgitation. It also showed the first signs of ascending aortic dilation. He remained symptomatic with breathlessness, particularly when feeding. At 5 months of age cardiac catheterisation for persistent pulmonary hypertension was performed to exclude a shunt. It indicated increased pulmonary vascular resistance that could not be accounted for by his cardiac disease alone. Furthermore, a chest radiograph performed during this procedure appeared to show an elevated left hemidiaphragm which had not been present on previous chest x-rays (figure 1). The following day this was confirmed on a CT scan, revealing eventration of the left hemidiaphragm and a right anterior diaphragmatic hernia (Morgagni hernia) with a small amount of liver herniating into the thoracic cavity (figure 2). At 6 months of age he had a laparotomy and repair of the right diaphragmatic hernia and plication of the left diaphragm. He remained stable postoperatively and was discharged home.

Chest x-ray showing an elevated left hemidiaphragm.
{kind=link}
{kind=link}
CT showing the right diaphragmatic hernia with the right lobe of the liver herniating into the thoracic cavity (see arrow).
Investigations
ECHO on day 1 of life: persistent pulmonary hypertension in the setting of a dysplastic tricuspid valve with severe regurgitation, biventricular hypertrophy and a PFO was reported.
Chest x-ray: A chest x-ray showing an elevated left hemidiaphragm (figure 1).
CT of the chest and abdomen: There is a 2 cm diameter deficiency of the right hemidiaphragm anteriorly in the parasternal area allowing the part of the right lobe of the liver to herniate into the chest abutting the right ventricle contributing to abnormality seen at the cardiophrenic angle. The left hemidiaphragm is elevated with bowel loops in the upper chest owing to eventration of the left hemidiaphragm (figure 2).
Genetics:
-
MFS: c.5202delA mutation in exon 41 of the FBN1 gene.
-
DS: chromosomal analysis confirmed trisomy 21.
Outcome and follow-up
On follow-up at 12 months of age he remained asymptomatic with no evidence of demonstrable pulmonary hypertension or diaphragmatic hernia. He continued to have moderate tricuspid regurgitation on ECHO for which he is under regular cardiac follow-up. His developmental milestones were delayed (at 12 months of age his development is equivalent to a 6-month old) but this was in keeping with his trisomy 21 and recurrent hospital admissions. He is under ongoing follow-up with the community paediatric team.
Discussion
DS, caused by trisomy 21, is the most common chromosomal abnormality and its phenotypic expression is well recognised in neonates. Its prevalence is 1–2/1000 live-births. MFS is caused by a mutation in the FBN1 gene on chromosome 15. Its estimated prevalence is 1/3000–5000 live-births.1 The expression of MFS is variable and it is estimated in only 14% of the cases present in the neonatal period.2 Neonatal MFS is thought to represent a genuine clinical entity with severe valvular heart lesions and congenital emphysema being predominant features.3
There is only one previous case report of the co-occurrence of DS and MFS in a 28-year-old woman.4 In this case, it was postulated that the phenotypic expression of MFS had been masked by DS. In our case, the phenotypic expression of DS had been masked by MFS; the only clinical features suggesting a diagnosis of DS were hypotonia and small ears. Other common clinical features, such as short fingers and short, broad hands were not seen as in this case the baby had large hands and feet with arachnodactyly secondary to MFS. Interaction between the two syndromes means that certain typical phenotypic features of DS may be diminished.
Conversely, there are certain clinical features that are common to the two conditions. With regard to congenital cardiac anomalies, neonatal MFS is commonly associated with severe valvular heart lesions as well as aortic root dilatation. DS is more commonly associated with atrioventricular septal defects; however, it has also been associated with atrioventricular valve abnormalities as shown in some case series.5 Furthermore, children with DS have a greater risk of pulmonary artery hypertension as present in this case.6
Congenital diaphragmatic hernia (CDH) is usually an isolated non-syndromic presentation, but it has been associated with a number of genetic lesions, the most common of which are chromosomal duplications.7 In one study, 15% of children with Morgagni hernia, as in this case, had DS.8 In addition, studies have shown that the diagnosis of Morgagni hernia can be delayed in children with DS.9 CDH has also been reported in syndromes caused by mutation in a specific gene including neonatal MFS, although evidence of this are isolated to occasional case reports.
In conclusion, we postulate that the phenotypic features of DS were masked by MFS, leading to the atypical presentation and delayed diagnosis. In addition, there is interaction and overlap of the two syndromes resulting in congenital cardiac abnormalities and congenital diaphragmatic hernia. We hope future reports of DS with other clinical entities will help to further understand its interaction with other syndromes.
Learning points
-
Down syndrome (DS) should be considered in the differential diagnosis of neonates with hypotonia.
-
Masking of DS phenotypic features can occur when associated with another syndrome.
-
As DS is so common it is important to consider its co-occurrence when certain clinical features are not in keeping with another known clinical syndrome.
-
There appears to be an overlap of symptoms and features between the syndromes. For example both DS and Marfan syndrome can cause congenital cardiac anomalies and have been associated with congenital diaphragmatic hernia.
Footnotes
-
Competing interests None.
-
Patient consent Obtained.
-
Provenance and peer review Not commissioned; externally peer reviewed.