
Abstract
Cardiac injury in patients infected with the novel Coronavirus (COVID-19) seems to be associated with higher morbimortality. We provide a broad review of the clinical evolution of COVID-19, emphasizing its impact and implications on the cardiovascular system. The pathophysiology of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) is characterized by overproduction of inflammatory cytokines (IL-6 and TNF-α) leading to systemic inflammation and multiple organ dysfunction syndrome, acutely affecting the cardiovascular system. Hypertension (56.6%) and diabetes (33.8%) are the most prevalent comorbidities among individuals with COVID-19, who require hospitalization. Furthermore, cardiac injury, defined as elevated us-troponin I, significantly relates to inflammation biomarkers (IL-6 and C-reactive protein (CRP), hyperferritinemia, and leukocytosis), portraying an important correlation between myocardial injury and inflammatory hyperactivity triggered by viral infection. Increased risk for myocardial infarction, fulminant myocarditis rapidly evolving with depressed systolic left ventricle function, arrhythmias, venous thromboembolism, and cardiomyopathies mimicking STEMI presentations are the most prevalent cardiovascular complications described in patients with COVID-19. Moreover, SARS-CoV-2 tropism and interaction with the RAAS system, through ACE2 receptor, possibly enhances inflammation response and cardiac aggression, leading to imperative concerns about the use of ACEi and ARBs in infected patients. Cardiovascular implications result in a worse prognosis in patients with COVID-19, emphasizing the importance of precocious detection and implementation of optimal therapeutic strategies.
Similar content being viewed by others
Introduction
Currently, the world is facing a pandemic caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), etiological agent of COVID-19 disease [1]. The first signs of the new virus began to show up in December 2019 in China. In this century, two coronavirus epidemics occurred, the severe acute respiratory syndrome coronavirus (SARS-CoV) in 2002 and the Middle East respiratory syndrome coronavirus (MERS-CoV) in 2012 [1]. SARS-CoV-2 presents a transmissibility ten times faster than 2002 SARS-CoV [1]. Due to its fast dissemination, COVID-19 was declared a pandemic by the World Health Organization (WHO). By the end of June 2020, the world had already registered more than 9 million individuals contaminated, with more than 450,000 deaths.
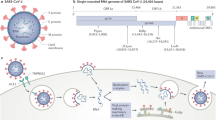
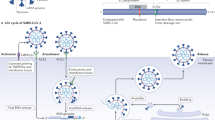
The coronaviruses are single-stranded RNA viruses, with great capacity for fast mutations and recombination, causing respiratory or intestinal infections in humans and animals [2]. The SARS-CoV-2 infection occurs through the coupling of S-protein located on the surface of the virus with angiotensin-converting enzyme 2 (ACE2), which acts as a receptor for the virus. ACE2 is mostly present in the lungs and seems to be the main gateway for the virus. It is also present in great amounts in the heart, which can lead to cardiovascular (CV) complications [2].
The main clinical manifestations of COVID-19 are respiratory, varying from a mild presentation to acute respiratory distress syndrome (ARDS), being potentially fatal. Moreover, as in other respiratory infections, pre-existing CV diseases and risk factors can increase the severity of COVID-19, leading to the aggravation and decompensation of chronic underlying cardiac pathologies as well as acute-onset of new cardiac complications [3], highlighting that myocardial injury can be present in approximately 12% of hospitalized patients with SARS-CoV-2 infection [1].
Within the CV manifestations of COVID-19, we can highlight four different aspects: (a) CV risk factors and established CV disease is associated with a worse prognosis, (b) appearance of acute CV complications in previously healthy individuals, (c) promising therapies with antimalarials and antivirals present important CV side effects, and (d) questioning the safety of the use of renin–angiotensin–aldosterone system (RAAS) inhibitors regarding an increased risk of COVID-19 [1]. Thus, the need to elucidate the potential pathophysiological mechanisms caused by COVID-19 and its CV repercussions becomes evident.
Methodology
We performed a thorough literature review based on the PubMed electronic bibliographic database. The following descriptors were used in the search engines advanced tool: “COVID-19,” “2019-nCoV,” “SARS-CoV-2,” “Myocardial Infarction,” “Coagulation,” “Heart Failure,” “Cardiovascular Risk Factors,” “Hypertension,” “Arrhythmias,” “Myocarditis.” Authors included retrospective studies, prospective studies, meta-analysis, and clinical guidelines regarding the cardiovascular implications related to COVID-19. Articles with a central theme diverging or not related to the cardiovascular complications associated with SARS-CoV-2 infection were excluded from the revision. Articles that were not in English were also excluded. A total of 35 citations were thoroughly reviewed to compose the present article. Preprints were included.
Discussion
Clinical evolution
The symptoms and clinical evolution of COVID-19 are broad, ranging from asymptomatic to severe respiratory failure requiring mechanical ventilation [3]. Therefore, to better understand the pathophysiology of the CV system injury, we will detail the clinical evolution of the disease.
The first, or “mild,” phase, commonly of benign evolution, occurs in the first 7 days with symptoms characteristic of upper respiratory tract infection. Nonspecific symptoms such as anosmia, ageusia, and gastrointestinal manifestations may also appear. Approximately 80% of the cases are resolved [3].
The second phase, or moderate pneumonia, occurs approximately in 15% of the patients and from the tenth day the symptoms begin to worsen with dyspnea, cough, and oxygen saturation decrease [3], with signs and symptoms suggesting progession for lower respiratory tract infection. Laboratory evaluation reveals an increase of inflammatory reagents such as C-reactive protein (CRP), ferritin, and D-dimer, evidencing an important inflammatory activity and a prothrombotic component of the disease. Additionally, a chest computed tomography (CT) may demonstrate a bilateral peripheral pulmonary infiltrate in a well-defined ground-glass pattern, indicating the development of viral pneumonia [3].
About 5% evolve to the third phase, or severe pneumonia, with worsening of the respiratory condition, hypoxemia, and fever [3]. In a pathophysiological standpoint, this phase is characterized by hyperactivity of inflammatory immune response, promoting a cytokinetic storm, resulting in hypercytokinaemia, mainly at the expense of interleukin 6 and 2 (IL-6 and IL-2), and tumor necrosis factor-α (TNF-α) [4]. Hypercytokinemia causes endothelial damage and tissue factor exposure with consequent activation of the coagulation cascade, generating a state of hypercoagulability, thromboembolic events, and increased risk of bleeding by disseminated intravascular coagulation (DIC). In addition, this phase is characterized by cytopenias and increased D-dimer [4].
Respiratory failure advances to ARDS. In chest CT, there is an exacerbation of the bilateral pulmanary ground-glass opacities with possible concomitant foci of consolidation [3]. Another severe complication is sepsis due to viral dissemination and inflammatory hyperactivity, progressing to multiple organ dysfunction syndrome.
It is noteworthy that the systemic inflammation promoted by SARS-CoV-2 plus hypercytokinaemia and hypercoagulability results in myocardial injury with increase of troponin and N-terminal prohormone of brain natriuretic peptide (NT-proBNP), and consequently CV complications, which will be further clarified below [4].
Cardiovascular complications
Cardiovascular risk factors
Hypertension and diabetes are the most prevalent comorbidities among individuals with COVID-19 requiring hospitalization. In a case-series study, 5,700 patients (average age of 63 years; 39.7% female) were included, being the most frequent comorbidities hypertension (56.6%), obesity (41.7%), and diabetes (33.8%) [5]. In general, recent studies demonstrate that patients with COVID-19 and hypertension have a higher mortality risk when compared with non-hypertensives [6].
It is worth mentioning that these patients are already naturally inflamed, where the association with the severe inflammatory process caused by the virus leads to an imbalance between the infection-induced increase in metabolic demand and the decrease in cardiac reserve [1, 4, 6]. On the other hand, we have to remember that the patients with the worst evolution are the elderly, where the prevalence of hypertension and diabetes is higher.
Hypertension, as a chronic illness, implies a proinflammatory state, as seen in several infectious disorders, as well as the attenuation of the innate immune response, being an important component of the COVID-19 pathogenesis, particularly in critical patients. Furthermore, new evidence is surfacing linking hypertension to a higher risk of developing severe disease due to SARS-CoV-2 infection [7].
A retrospective cohort study included 126 patients with COVID-19 and pre-existent hypertension, and 125 age- and sex-matched patients with COVID-19 without hypertension. Among hypertensives, 21.3% had more severe SARS-CoV-2 infection and higher death rate, 10.3% vs. 6.4%, when compared with the non-hypertension group, indicating that hypertension is a critical risk factor associated with poorer clinical outcomes. Also, hypertensives with COVID-19 presented higher concentrations of high-sensitivity CRP, procalcitonin, and IL-6 when compared to controls, indicating that hypertension enhances inflammation in SARS-CoV-2 infection. Interestingly, hypertensive patients undergoing anti-hypertensive therapy with RAAS blockers presented a less profound inflammatory profile when compared with non-ARBs/ACEi treatment [8].
Hypertension seems to enhance the inflammatory profile in patients with SARS-CoV-2 infection, inferring that resistant hypertension possibly increases even more the risk for a more severe illness, as it was already demonstrated that these patients present higher levels of inflammatory biomarkers such as TNF-α and IL-6; however, more evidence is still necessary to elucidate the potential pathophysiological relationship, clinical evolution and eventual risk of patients with resistant hypertension and COVID-19 [9].
Myocarditis
The SARS-CoV-2 infection, mainly in its severe forms, apparently induce an aggression to the myocardium resulting in myocarditis [1]. Shi et al. [10] reported that patients with myocardial injury, defined by the increase of troponin levels, presented mortality greater than those without myocardial injury (51.2% vs. 4.5%; p < 0.001), being an independent risk factor for mortality [10]. In addition, myocardial injury was associated with a higher incidence of ARDS (58.5% vs. 14.7%), higher need of non-invasive (46.3% vs. 3.9%) or invasive ventilation (22.0% vs. 4.2%), and complications such as acute kidney injury (8.5% vs. 0.3%) and coagulopathy (7.3% vs. 1.8%) [10]. Thus, troponin seems to be a marker of worse prognosis and mortality in patients with COVID-19. In addition, patients with an increase of troponin present higher levels of leukocytes, D-dimer, CRP, ferritin, and IL-6, portraying an important correlation between myocardial injury and inflammatory hyperactivity triggered by the viral infection [10].
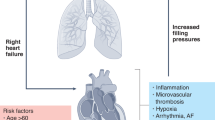
The pathophysiology regarding myocardial damage caused by COVID-19 infection is not completely elucidated. However, studies highlight that the pathogenesis may reflect a process of viral replication and dissemination within the cardiomyocytes themselves. Moreover, it should be noted that hypercytokinaemia triggered by SARS-CoV-2 with consequent systemic inflammation may induce myocardial damage [4, 11]. Based mainly on experimental studies in animals, the lower expression of ACE2 in the myocardial tissue resulting in tissue inflammation is also a hypothesis [12]. Therefore, the consequent myocardial inflammation may result in an abrupt decrease of the cardiac contractility strength, generating inotropic deficit, increased filling pressures, and acute heart failure.
Myocarditis related to SARS-CoV-2 has been described as an important acute ventricular dysfunction associated with diffuse myocardial edema [13]. Furthermore, it is important to accentuate the existence of reports revealing the occurrence of myocarditis with fulminant evolution, complicated with pericarditis, pericardial effusion, and consequent cardiac tamponade [13]. Inciardi et al. [11] emphasized that the myocardial involvement may occur even in the absence of symptoms of upper respiratory tract infection.
On physical examination, the presence of hypotension, tachycardia, tachypnea, signs of low cardiac output, and a third heart sound have been reported. It is noteworthy that the electrocardiogram (ECG) may show diffuse ST segment elevation with concave morphology in association with a significant increase in troponin, brain natriuretic peptide (BNP)/NT-proBNP, and evidence of inflammatory activity [11]. Also, transthoracic echocardiography can demonstrate diffuse hypokinesia with myocardial thickening and decrease in the left ventricular ejection fraction, while cardiac magnetic resonance imaging reveals diffuse interstitial edema [11]. It is important to keep in mind that all of these complementary exams have important restrictions requiring its own logistics due to the high contagion power of SARS-CoV-2 [1]. From a therapeutic standpoint, patients with clinical signs of tissue hypoperfusion and fluid overload have been submitted to inotropic support and diuretic therapy [11]. In addition, circulatory assist devices such as venoarterial extracorporeal membrane oxygenation (ECMO) have been used in patients with cardiogenic shock and severe myocardial injury with hemodynamic impairment [1, 10, 11, 13].
Myocardial injury is a factor of worse prognosis and is directly associated with a higher mortality in COVID-19. Thus, it is imperative the implementation of a thorough screening through dosage of troponin, ECG, and bedside echocardiography, mainly in patients presenting signs of greater severity and critical infection, due to the association with exacerbated systemic inflammation [10,11,12].
Heart failure
Heart failure is an important cause of death in patients with COVID-19 and occurs as a result of different myocardial aggression mechanisms such as direct myocardial injury by viral action, indirect and direct inflammatory damage, O2 supply–demand imbalance, and increase of atherothrombotic events due to inflammatory destabilization of atheromatous plaques resulting in acute myocardial dysfunction [11, 14].
Direct myocardial damage occurs as a result of SARS-CoV-2 interaction with myocardial tissue by binding the viral glycoprotein Spike 1, after its activation by the serine 2 transmembrane protease expressed by the host to ACE2 receptors, expressed especially in cardiac pericytes, leading to direct tissue damage and, in a further moment, a downregulation of these receptors [14].
Guo et al. [14] reported that the increase of serum troponin in patients with or without previous CV disease has been directly related with an increase of plasmatic NT-proBNP levels, increasing mortality, being characterized as a marker of poor prognosis. Nonetheless, the increase of troponin in individuals with previous CV disease characterized more severe clinical outcomes and higher mortality when compared to patients without previous heart disease and increased troponin values [11, 14]. Hence, the presence of heart disease is an important risk factor regarding the prognosis of COVID-19 patients evolving with acute myocardial injury [10, 14].
The hypothesis of the cumulative effect of previous CV disease and troponin increase was postulated due to the greater presence of ACE2 receptors in postmortem cardiac pericytes extracted from patients with heart disease compared with those without previous disease [15].
Acute myocardial infarction
Despite the level of scientific evidence on the subject is still low, there are many studies correlating acute myocardial injury (AMI) and the infection by SARS-CoV-2 [10, 16]. According to the Fourth Universal Definition of AMI, it can be defined in the presence of AMI with clinical or laboratorial evidence of myocardial ischemia, being divided in five different pathophysiological types [16].
Some of the hypotheses raised so far may clarify the relationship between COVID-19 and ischemic events [4]. Together with the exacerbated inflammation, other factors such as hypoxemia, immobilization, and, in some cases DIC, culminate in a prothrombotic state. Therefore, it can precipitate type-1 AMI, related to instability of the atherosclerotic plaque, leading to coronary thrombosis and subsequent infarction [16, 17]. Nevertheless , when compared to patients hospitalized with severe pneumonia due to other etiologies, individuals infected with SARS-CoV-2 have greater thrombophilia, which raises questions about the other mechanisms [17].
Bioinformatics studies suggested that some binding proteins and viral envelope glycoproteins may bind to both porphyrin and the β-chain of hemoglobin. This association may lead to a decrease of available serum hemoglobin and, consequently, to hypoxemia, with pulmonary and cardiac consequences. The inability to perform gas exchange may precipitate an intense inflammation of lung tissues [18]. Type-2 AMI, which occurs due to a myocardial oxygen demand disproportionate to the supply, is explained by hypoxemia associated with increased cardiac demand due to systemic infection. It is estimated that ~7% of patients have myocardial ischemia or myocarditis [16].
In addition, it is important to note the possibility of a last adverse mechanism. Patients previously submitted to angioplasty may, due to the hypercoagulability state induced by the infection, present an increased incidence of type-4b AMI due to thrombotic occlusion of stent [16, 17].
Takotsubo cardiomyopathy
Takotsubo cardiomyopathy and its reverse presentation are important differential diagnosis of myocardial injury. So far, there are some very heterogeneous case reports, showing laboratory and ECG changes typical of myocardial injury, such as increased troponin, NTPro-BNP, T-wave inversion, and ST segment elevation, in addition to transthoracic echocardiogram revealing left ventricular dysfunction, associated with hyperkinesia in apical ballooning, characteristic of Takotsubo cardiomyopathy [19].
It is important to note that the current pandemic scenario may represent a potential trigger for Takotsubo cardiomyopathy, not only due to the respiratory infection, but by the profound emotional stress caused by the isolation period leading to an excessive release of catecholamines [19].
Venous thromboembolism
There is evidence that infection by COVID-19 predisposes to a prothrombotic state by direct mechanisms (microvasculitis due to viral damage), indirect mechanisms (downregulation of ACE2 receptor, hypoxia, and DIC), and even behavioral mechanisms (bed restriction due to prolonged mechanical ventilation and decreased deambulation due to social isolation) [17, 20]. According to recent studies, this prothrombotic state could increase the risk of arterial thrombosis but also of venous thromboembolism, such as deep vein thrombosis (DVT) and pulmonary thromboembolism (PTE) described in patients with severe infection by SARS-CoV-2 [20]. This pathophysiological mechanism is supported by the high values of D-dimer in patients with PTE and COVID-19, which presented a high sensitivity and specificity for the presence of thrombi in chest angiotomography [20].
A Chinese study with 449 patients, of whom 99 used low-molecular-weight heparin for at least 7 days, demonstrated that D-dimer levels, prothrombin time, and age were positively associated with a 28-day mortality. The mortality was similar between those who used or did not use heparin, but it was lower among those who used heparin in the subgroup with D-dimer six times greater than the upper limit (40.0% vs. 64.2%, p = 0.029) and with sepsis-induced coagulopathy score ≥ 4 (32.8% vs. 52.4%, p = 0.017) [21]. Thus, the anticoagulant treatment in patients with severe COVID-19 based on serum D-dimer levels has been preconized [21].
Arrhythmias
There are few reports in the literature regarding cardiac arrhythmias in COVID-19, with no clear pathophysiological correlation. The potential mechanism seems to be viral myocarditis caused by SARS-CoV-2 [19]. Fulminant myocarditis associated with cardiogenic shock may be associated with the development of both ventricular and atrial arrhythmias, as myocardial inflammation itself with severe necrosis may develop re-entry points in the electrical circuit, evolving with ventricular tachycardia and ventricular fibrillation [22, 23].
Supporting this hypothesis, some reports describe a strong correlation between high troponin levels and increased CRP with a higher incidence of ventricular arrhythmias and greater need of mechanical ventilation [22]. Guo et al. [14] also correlated the increase in NT-ProBNP levels with the onset of malignant arrhythmias, suggesting the relationship between acute myocarditis and arrhythmias. However, it is necessary to keep in mind that these patients who evolve with a severe inflammatory phase develop severe hypoxemia and hydroelectrolytic disorder, causing arrhythmias potentially related to potassium disorders [22]. Moreover, transient complete atrioventricular block was also described in critical patient, raising the possibility of myocarditis or due to increase of pulmonary artery pressure [22, 23].
Antimalarial and antiviral use during COVID-19 pandemic: impact on the cardiovascular system
The ECG plays a key role in monitoring the corrected QT interval (QTc) in hospitalized patients using hydroxychloroquine (HCQ) and azithromycin. It is noteworthy that this drug association is being used, by some centers, in hospitalized patients because of their potential ability to interfere in the cell endocytosis of the virus. However, both drugs are correlated with the prolongation of the QT interval and consequently with an arrhythmogenic potential for malignant ventricular arrhythmias, especially polymorphic ventricular tachycardia with enlarged QT interval (Torsades des Pointes) [23]. It is worth noting that patients who already present borderline QT interval have a greater risk of developing ventricular arrhythmia.
A multicenter, open-label, randomized controlled trial of 150 patients with mild–moderate COVID-19 revealed that antimalarial drugs led to a similar probability of virus eradication when compared to standard care, establishing neither benefits nor significant increase of risks, discouraging the use of HCQ in those patients [24]. However, in a cohort study of 90 hospitalized patients with COVID-19, patients taking HCQ and azithromycin had greater QT interval prolongation than those taking HCQ alone [25].
Macrolides, such as azithromycin, are antibiotics with immunomodulatory and anti-inflammatory effects that also carry the risk of QT interval prolongation and/or torsade des pointes [26]. In a pathophysiological perspective, it is known that both antimalarial drugs and macrolides produce a QT interval prolongation via a blockade of the rapidly activating delayed rectifier potassium current (IKr), encoded by the human-ether-a-go-go-related gene, causing a delay in phase 3 rapid repolarization of the action potential. This mechanism lengthens ventricular repolarization and the duration of ventricular action potential, leading to after depolarizations, due to activation of depolarizing currents, and resultant ventricular extrasystoles. Moreover, recurring ventricular extrasystoles associated with areas of slow conduction optimizes electrical re-entry circuits, increasing the risk of torsades des pointes and sudden cardiac death [27].
Antiviral drugs, such as remdesivir, lopinavir/ritonavir, and ribavirin, are also being administered in patients with more severe COVID-19 due to their potential benefit in viral RNA synthesis inhibition, diminishing RNA translation and viral exocytosis in host cells. In a cohort of 61 patients who received remdesivir therapy, 12% of the patients evolved with hypotension, being one of the most common serious adverse event, particularly in patients undergoing invasive ventilation. The improvement in oxygen support status was observed in 68% of the patients in the remdesivir group [28].
Ribavirin and lopinavir/ritonavir are also under investigation in numerous clinical trials for COVID-19 and they also may result in QT interval prolongation, through IKr blockade, especially in patients with baseline QTc abnormality. In a randomized, controlled, open-label trial with 199 patients, investigating lopinavir/ritonavir efficacy for SARS-CoV-2 infection, only 1.1% of the patients in the lopinavir/ritonavir group developed QT interval prolongation and no clinical benefit was observed with lopinavir/ritonavir treatment beyond standard care [20]. Furthermore, lopinavir/ritonavir can affect anticoagulant therapy pharmacodynamics, making it important to emphasize that critical SARS-CoV-2 can be associated with hypercoagulability and a prothrombotic state [28, 29].
RAAS and SARS-CoV-2
The interaction between the virus SARS-CoV-2 and RAAS has been proposed as a potential factor in the infectivity of patients, causing several concerns about the use of RAAS inhibitors that may be linked to the action of ACE2, possibly responsible for the virulence of the disease. We know that these drugs are among the most commonly prescribed for the treatment of hypertension, diabetes, and heart failure worldwide, clinical conditions that are strongly associated with an increased risk of an unfavorable evolution of COVID-19 [30].
Some studies suggest that the cardiac damage secondary to SARS-CoV-2 may be linked to ACE2, present in the cell membrane of CV, lung, and kidney tissues. The physiological role of ACE2 is to perform the conversion of angiotensin II in a molecule of angiotensin 1–7, causing vasodilation and decrease of hydric retention, through Mas receptor, acting as counter-regulator of the blood pressure elevation [31].
Preliminary studies showed that SARS-CoV-2, through its surface protein Spike, is capable of binding to ACE2-anchoring receptor in host cells, facilitating its entry in cardiac and lung cells, specially deregulating RAAS and causing a downregulation effect of ACE2, which leads to the accumulation of angiotensin II with proinflammatory effect. The SARS-CoV-2 surface features the multiple spike glycoproteins (S) displayed in a crown-like appearance (corona) which, after being primed by the host’s TMPRSS2 membrane protease, are used by the virus to engage its main internalization receptor: ACE2, the same functional receptor of the earlier SARS-CoV [2].
Nevertheless, despite preserving 8 of the 14 amino acid residues that make up the coronavirus binding domain of the spike protein of SARS-CoV, a three-dimensional analysis of the receptor-binding domain of SARS-CoV-2-binding site reveals that enhanced ACE2 receptor-binding affinity, along with the presence of a novel polybasic furin cleavage site inserted at the boundary of the S1/S2 subunits of the spike S-protein, may account for the increased infectivity and virulence of the SARS-CoV-2 in comparison with SARS-CoV [32,33,34]. Studies demonstrate that ACE2 deactivation may play a harmful role in the development of respiratory failure and its progression [30]. It is important to highlight that, despite the structural homology between ACE2 and ACE, its enzymatic active sites are different and, as a result, ACEi in clinical use do not directly affect ACE2 activity. In addition, ACE2 plays a well-known role in the ventricular function recovery of patients with myocardial injury, because it inhibits angiotensin II activity [30, 35].
Likewise, individuals with hypertension would have a higher expression of ACE2 secondary to the use of RAAS inhibitors, which would potentially increase the susceptibility to infection by SARS-CoV-2. In animal models, the use of these drugs can generate an upregulation effect, producing a greater number of ACE2 receptors in the myocardium and lung cells, inferring a facilitated entry of the virus into the host cells consequently enhancing SARS-CoV-2 virulence [30, 35]. Nonetheless, evidences initially deducing that the use of RAAS blockers during the COVID-19 might be deleterious were not confirmed.
In a large population-based case–control study, the impact of RAAS blockers use on COVID-19 severity was assessed in 6,272 patients who tested positive for SARS-CoV-2 infection; the use of ACEi/ARBs was not associated with an increased COVID-19 risk or a worse prognosis [30]. Likewise, in a retrospective study with 12,594 individuals evaluating the relation between previous hypertension treatment with ACEi, ARBs, β-blockers, calcium-channel blockers, or thiazide diuretics and increased severe illness and contamination risk, no substantial association between the use of these drugs and more severe SARS-CoV-2 infection was demonstrated [33].
Therefore, ACEi/ARBs are possibly potential drugs for COVID-19 treatment in patients with pre-existing hypertension, due to their capacity of reducing the hyperinflammatory state particularly observed in patients with critical SARS-CoV-2, although a less significant reduction in 1L-6 serum levels suggests that RAAS blockers alone might be inefficient towards inflammation control [8]. Hence, authors are postulating a possible beneficial synergic effect between a ACEi/ARB combination with IL-6R inhibitors towards inflammation attenuation in patients with hypertension and COVID-19, requiring more robust evidence through randomized controlled trials [8].
Initial concern regarding RAAS blockers during the COVID-19 pandemic impelled international societies to issue positions advocating the importance of RAAS therapy maintenance in patients with formal indication during the pandemic. Current evidence indicates that hypertension per se might be a risk factor associated with a higher mortality and worse outcomes in patients with COVID-19, particularly patients with uncontrolled hypertension, and that controlled hypertension specifically with RAAS blocker therapy might be associated with more favorable outcomes.
Conclusion
Hypertension is strongly associated with SARS-CoV-2 infection possibly due to the proinflammatory state of this chronic illness in addition to the hypercytokinaemia that occurs in COVID-19. Myocardial injury is a prevalent complication in hospitalized patients, with or without previous CV disease, and presents a significant association with in-hospital mortality and a worse evolutive prognosis. It is worthy to highlight the importance of serial troponin dosage as a prognosis marker. In addition, myocardial insult was associated with severe ventricular dysfunction and higher incidence of ventricular arrhythmia, mainly evidenced through retrospective studies, emphasizing the importance of early detection and implementation of a proper therapy.
References
Clerkin KJ, Fried JA, Raikhelkar J, Sayer G, Griffin JM, Masoumi A, et al. Coronavirus disease 2019 (COVID-19) and cardiovascular disease. Circulation 2020;141:1648–55.
Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, et al. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181:271–80.
Wu Z, McGoogan JM. Characteristics of and important lessons from the coronavirus disease 2019 (COVID-19) outbreak in China. JAMA. 2020;323:1239–42.
Ye Q, Wang B, Mao J. The pathogenesis and treatment of the ‘Cytokine Storm′ in COVID-19. J Infect 2020;80:607–13.
Richardson S, Hirsch JS, Narasimhan M, Crawford JM, McGinn T, Davidson KW, et al. Presenting characteristics, comorbidities, and outcomes among 5700 patients hospitalized with COVID-19 in the New York City area. JAMA 2020;323:2052–9.
Zuin M, Rigatelli G, Zuliani G, Rigatelli A, Mazza A, Roncon L. Arterial hypertension and risk of death in patients with COVID-19 infection: systematic review and meta-analysis. J Infect 2020;81:e84–6.
Zheng A, Peng F, Xu B, Zhao J, Liu H, Peng J, et al. Risk factors of critical & mortal COVID-19 cases: a systematic literature review and meta-analysis. J Infect. 2020;S0163-4453:30234–6. https://doi.org/10.1016/.j.jinf2020.04.021.
Yang G, Tan Z, Zhou L, Yang M, Peng L, Liu J, et al. Effects of angiotensin II receptor blockers and ACE (angiotensin-converting enzyme) inhibitors on virus inhection, inflammatory status, and clinical outcomes in patients with COVID-19 and hypertension: a single-center retrospective study. Hypertension. 2020;76:51–8.
Barbaro NR, Fontana V, Modolo R, De Faria AP, Sabbatini AR, Fonseca F, et al. Increased arterial stiffness in resistant hypertension is associated with inflammatory biomarkers. Blood Press 2015;24:7–13.
Shi S, Qin M, Shen B, Cai Y, Liu T, Yang F et al. Association of Cardiac Injury With Mortality in Hospitalized Patients With COVID-19 in Wuhan, China. JAMA Cardiol. 2020;5:802–10. https://doi.org/10.1001/jamacardio.2020.0950.
Inciardi RM, Lupi L, Zaccone G, Italia L, Raffo M, Tomasoni D, et al. Cardiac Involvement in a Patient With Coronavirus Disease 2019 (COVID-19). JAMA Cardiol. 2020;5:1–6. https://doi.org/10.1001/jamacardio.2020.1096.
Guo J, Huang Z, Lv J. Coronavirus disease 2019 (COVID-19) and cardiovascular disease: a viewpoint on the potential influence of angiotensin-converting enzyme inhibitors/angiotensin receptor blockers on onset and severity of severe acute respiratory syndrome coronavirus 2 infection. J Am Heart Assoc 2020;9:e016219.
Hua A, O’Gallagher K, Sado D, Byrne J. Life-threatening cardiac tamponade complicating myo-pericarditis in COVID-19. Eur Heart J 2020;41:2130.
Guo T, Fan Y, Chen M, Wu X, Zhang L, He T et al. Cardiovascular Implications of Fatal Outcomes of Patients with Coronavirus Disease 2019 (COVID-19). JAMA Cardiol. 2020;5:1–8. https://doi.org/10.1001/jamacardio.2020.1017.
Chen L, Li X, Chen M, Feng Y, Xiong C. The ACE2 expression in human heart indicates new potential mechanism of heart injury among patients infected with SARS-CoV-2. Cardiovasc Res 2020;116:1097–100.
Thygesen K, Alpert JS, Jaffe AS, Chairman BR, Bax JJ, Morrow DA, et al. Executive group on behalf of the Joint European Society of Cardiology (ESC)/American College of Cardiology (ACC)/American Heart Association (AHA)/World Heart Federation (WHF) Task Force for the Universal Definition of Myocardial Infarction. Fourth Univers Défin Myocard Infarct Glob Heart 2018;13:305–38.
Klok FA, Kruip MJHA, van der Meer NJM, Arbous MS, Gommers DAMPJ, Kant KM, et al. Incidence of thrombotic complications in critically ill ICU patients with COVID-19. Thromb Res 2020;191:145–7.
Wenzhong L, Hualan L. COVID-19: attacks the 1-beta chain of hemoglobin and captures the porphyrin to inhibit human heme metabolism. ChemRxiv. Preprint at https://doi.org/10.26434/chemrxiv.11938173.v8.
Siripanthong B, Nazarian S, Muser D, Deo R, Santangeli P, Mohammed Y et al. Recognizing COVID-19-related myocarditis: the possible pathophysiology and proposed guideline for diagnosis and management. Heart Rhythm. 2020;S1547-5271:30422–7.
Bikdeli B, Madhavan MV, Jimenez D, Chuich T, Dreyfus I, Driggin E, et al. COVID-19 and thrombotic or thromboembolic disease: implications for prevention, antithrombotic therapy, and follow-up. J Am Coll Cardiol 2020;75:2950–73.
Tang N, Bai H, Chen X, Gong J, Li D, Sun Z. Anticoagulant treatment is associated with decreased mortality in severe coronavirus disease 2019 patients with coagulopathy. J Thromb Haemost 2020;18:1094–9.
Lakkireddy DR, Chung MK, Gopinathannair R, Patton KK, Gluckman TJ, Turagam M, et al. Guidance for cardiac electrophysiology during the coronavirus (COVID-19) pandemic from the Heart Rhythm Society COVID-19 Task Force; Electrophysiology Section of the American College of Cardiology; and the Electrocardiography and Arrhythmias Committee of the Council on Clinical Cardiology, American Heart Association. Heart Rhythm 2020; S1547-5271:30289–7.
Wu C-I, Postema PG, Arbelo E, Behr ER, Bezzina CR, Napolitano C, et al. SARS-CoV-2, COVID-19 and inherited arrhythmia syndromes. Heart Rhythm 2020;S1547-5271:30285-X.
Tang W, Cao Z, Han M, Wang Z, Chen J, Sun W, et al. Hydroxychloroquine in patients with mainly mild to moderate coronavirus disease 2019: open label, randomised controlled trial. BMJ 2020;369:m1849.
Mercuro NJ, Yen CF, Shim DJ, Maher TR, McCoy CM, Zimetbaum PJ, et al. Risk of QT interval prolongation associated with use of hydroxychloroquine with or without concomitant azithromycin among hospitalized patients testing positive for coronavirus disease 2019 (COVID-19). JAMA Cardiol. 2020;e201834. https://doi.org/10.1001/jamacardio.2020.1834
Hancox JC, Hasnain M, Vieweg WV, Crouse EL, Baranchuk A. Azithromycin, cardiovascular risks, QTc interval prolongation, torsade de pointes, and regulatory issues: a narrative review based on the study of case reports. Ther Adv Infect Dis 2013;1:155–65.
Traebert M, Dumotier B, Meister L, Hoffmann P, Dominguez-Estevez M, Suter W. Inhibition of hERG K+ currents by antimalarial drugs in stably transfected HEK293 cells. Eur J Pharmacol 2004;484:41–8.
Grein J, Ohmagari N, Shin D, Diaz G, Asperges E, Castagna A, et al. Compassionate use of remdesivir for patients with severe Covid-19. N Engl J Med 2020;382:2327–36.
Cao M, Wang Y, Wen D, Liu W, Wang J, Fan G, et al. A trial of Lopinavir-Ritonavir in adults hospitalized with severe Covid-19. N Engl J Med 2020;382:1787–99.
Mancia G, Rea F, Ludergnani M, Apolone G, Corrao G. Renin–angiotensin–aldosterone system blockers and the risk of Covid-19. N Engl J Med. 2020;382:2431–40.
Carey RM, Wang JG. Evidence that renin–angiotensin system inhibitors should not be discontinued due to the COVID-19 pandemic. Hypertension 2020;76:42–3.
Lan J, Ge J, Yu J, Shan S, Zhou H, Fan S, et al. Structure of the SARS-CoV-2 spike receptor-binding domain bound to the ACE2 receptor. Nature 2020;581:215–20.
Reynolds HR, Adhikari S, Pulgarin C, Troxel AB, Iturrate E, Johnson SB, et al. Renin-angiotensin-aldosterone system inhibitors and risk of Covid-19. N Engl J Med 2020;382:2441–8.
Zhang H, Penninger JM, Li Y, Zhong N, Slutsky AS. Angiotensin-converting enzyme 2 (ACE2) as a SARS-CoV-2 receptor: molecular mechanisms and potential therapeutic target. Intensive Care Med 2020;46:585–90.
Kreutz R, Algharably EAE, Azizi M, Dobrowolski P, Guzik T, Januszewicz A et al. Hypertension, the renin-angiotensin system, and the risk of lower respiratory tract infections and lung injury: implications for COVID-19. Cardiovasc Res. 2020. https://doi.org/10.1093/cvr/cvaa106.
Funding
This study was supported by research grants from the Conselho Brasileiro de Desenvolvimento Científico e Tecnológico (CNPq, Distrito Federal, Brazil) and Fundação Carlos Chagas Filho de Amparo à Pesquisa do Estado do Rio de Janeiro (FAPERJ, Brazil). The sponsors have no role in study design, data collection and analysis, results interpretation, or in preparation, review, and approval of the manuscript.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Azevedo, R.B., Botelho, B.G., Hollanda, J.V.G.d. et al. Covid-19 and the cardiovascular system: a comprehensive review. J Hum Hypertens 35, 4–11 (2021). https://doi.org/10.1038/s41371-020-0387-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41371-020-0387-4
This article is cited by
-
Incidence of new-onset hypertension before, during, and after the COVID-19 pandemic: a 7-year longitudinal cohort study in a large population
BMC Medicine (2024)
-
Cardiac deceleration capacity is associated with severity of inflammation in COVID-19
Infection (2024)
-
Identification of genetic loci jointly influencing COVID-19 and coronary heart diseases
Human Genomics (2023)
-
Role of OAS gene family in COVID-19 induced heart failure
Journal of Translational Medicine (2023)
-
Impact of Covid-19 on risk of severe maternal morbidity
Critical Care (2023)
