Article Text

Summary
Hydralazine, a vasodilator, is commonly used as an adjunctive treatment for moderate to severe hypertension, heart failure and hypertensive emergencies in pregnancy. Hydralazine-induced lupus was first described in 1953. Clinical presentation ranges from arthralgia, myalgia, petechiae, or rash to single or multiorgan involvement. An occurrence of systemic vasculitis is a rare complication. When presented as the pulmonary–renal syndrome, it could have a rapidly progressive course which can be fatal. Here, we describe a case of hydralazine-associated rapidly progressive glomerulonephritis and pulmonary haemorrhage. We use this case to review the current literature and discuss and highlight the importance of a high degree of clinical acumen, early diagnosis and prompt treatment for better clinical outcomes.
- drug interactions
- renal system
- acute renal failure
- vasculitis
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Background
Hydralazine was discovered in the 1950s as a treatment for malaria. It is a direct smooth muscle vasodilator used as an adjunctive treatment for moderate to severe hypertension, heart failure with reduced ejection fraction (ACE inhibitor or angiotensin receptor blocker intolerance) and in hypertensive emergencies in pregnancy.1 Hydralazine is often implicated as a causal factor in drug-induced lupus. Hydralazine-induced antinuclear cytoplasmic antibody (ANCA) vasculitis (AAV) is a rare phenomenon with pulmonary–renal syndrome with the most severe presentation. The pathogenesis of hydralazine-associated AAV is unknown. Theories for the mechanism of hydralazine-associated AAV include,2 (1) hydralazine binding to myeloperoxidase (MPO) leading to neutrophil apoptosis, resulting in the production of multiple autoantibodies; (2) hydralazine-induced reversal of epigenic silencing of MPO and proteinase3 (PR3) leading to increased expression of neutrophil autoantigens and (3) a break in tolerance in slow versus fast acetylators of hydralazine.3 Here, we report a severe complication of hydralazine, presenting as rapidly progressive glomerulonephritis (GN) with pulmonary haemorrhage and discuss the current literature.
Case presentation
A 65-year-old Caucasian woman with a prior history of bilateral clear cell renal cell carcinoma (2013) treated with bilateral open partial nephrectomies, poorly controlled diabetes mellitus over 30 years, hypertension, obesity with prior gastric bypass surgery, progressive proteinuric chronic kidney disease stage 4, presented to the hospital from home with a 2 day history of shortness of breath and lower extremity swelling and weakness. On presentation, she was afebrile with a blood pressure of 178/88 mm Hg, heart rate of 84 beats/min, pulse oxygenation of 95% on 2 L of O2. The physical was notable for an obese woman with tachypnoea, fine rales and 1+pitting pedal oedema. Initial laboratory workup showed a serum creatinine of 6.1 mg/dL with an estimated glomerular filtration rate of 8 mL/min/1.73 m2(eGFR) (calculated using the four-variable modification of diet in renal disease (MDRD) study equation), blood urea nitrogen of 74 mg/dL, potassium of 3.6 mmol/L, haemoglobin of 7 g/dL, white blood cell count of 6.9 x109/L and platelets of 141 x109/L.A urine analysis showed 300 protein, specific gravity of 1.020, large blood and >100 of red blood cells (RBC)/hpf. Chest X-ray was notable for central vascular congestion. A renal ultrasound showed smaller sized kidneys with no hydronephrosis, calculi, with mild to moderate diffuse parenchymal thinning. Her home medications included atorvastatin, bupropion, aspirin, calcitriol, furosemide, hydralazine, Insulin glargine, levothyroxine, mirtazapine, pantoprazole and metoprolol. She was not a smoker with no history of alcohol use. Denied family history of renal disease.
A review of her outpatient laboratories 3 months prior showed a baseline creatinine of 2.4 mg/dL with corresponding eGFR of 21 mL/min/1.73 m2 (MDRD). Prior urinalysis was positive for 100 of protein with no RBC or blood. Prior serologies including Hepatitis B Ag, Hepatitis C Ab, HIV and antinuclear antibody (ANA) were all negative. Her spot urine protein creatinine ratio was 2.1 g.
Investigations
Serological work up, bronchoscopy, CT and renal biopsy.
Differential diagnosis
Acute renal failure with pulmonary oedema and uremic haemoptysis.
Acute heart failure.
Respiratory tract infection with prerenal renal failure and or postinfectious GN.
Systemic lupus erythematosus.
Cryoglobulinaemic vasculitis.
Treatment
The patient admitted and treated with gentle intravenous fluids and a unit of packed red blood cells. Hydralazine was stopped. Serological workup requested given active urine sediment along with a urological evaluation given the history of renal cell carcinoma. Her erythrocyte sedimentation rate was >100. Given the patient’s presentation, laboratory parameters with severe renal failure, urine sediment, high inflammatory markers, GN was high on the differential and pulse intravenous steroids administered while waiting for the final serologies and the renal biopsy. Perinuclear antinuclear cytoplasmic antibody titres were 1:40 (reference range <1:20, ARUP Laboratories, Utah, USA), MPO antibody was positive at 44 AU/mL (reference range 0–19) and serine protease 3 IgG was 5 AU/mL (reference range 0–19). ANA and glomerular basement membrane antibody were negative. Complement 3 was 72 mg/dL (reference range 88–201), Complement 4 was 17 mg/dL (reference range 10–40). Serum protein electrophoresis and Immunofixation electrophoresis were of the normal pattern. A renal biopsy performed.
The renal biopsy (figure 1) specimen contained 35 glomeruli, 17 of which were globally sclerotic and four were ischaemic. The remaining glomeruli were enlarged and with moderate to severe diffuse and global increase in mesangial matrix and nodule formation. Nine glomeruli had crescents, two of which were cellular, four of which were fibrocellular and three of which were fibrous. Three glomeruli exhibited segmental fibrinoid necrosis. Moderate tubular atrophy and interstitial fibrosis involving 40%–50% of cortical areas with patchy moderate interstitial inflammation by the lymphocytes, plasma cells and monocytes. Severe arteriosclerosis with hyalinosis observed. Immunofluorescence showed weak mesangial staining for IgG, IgA, C3, kappa and lambda, within the spectrum of pauci-immune necrotising crescentic GN. There were low levels of mesangial immune deposits. No definitive immune type electron dense deposits seen.

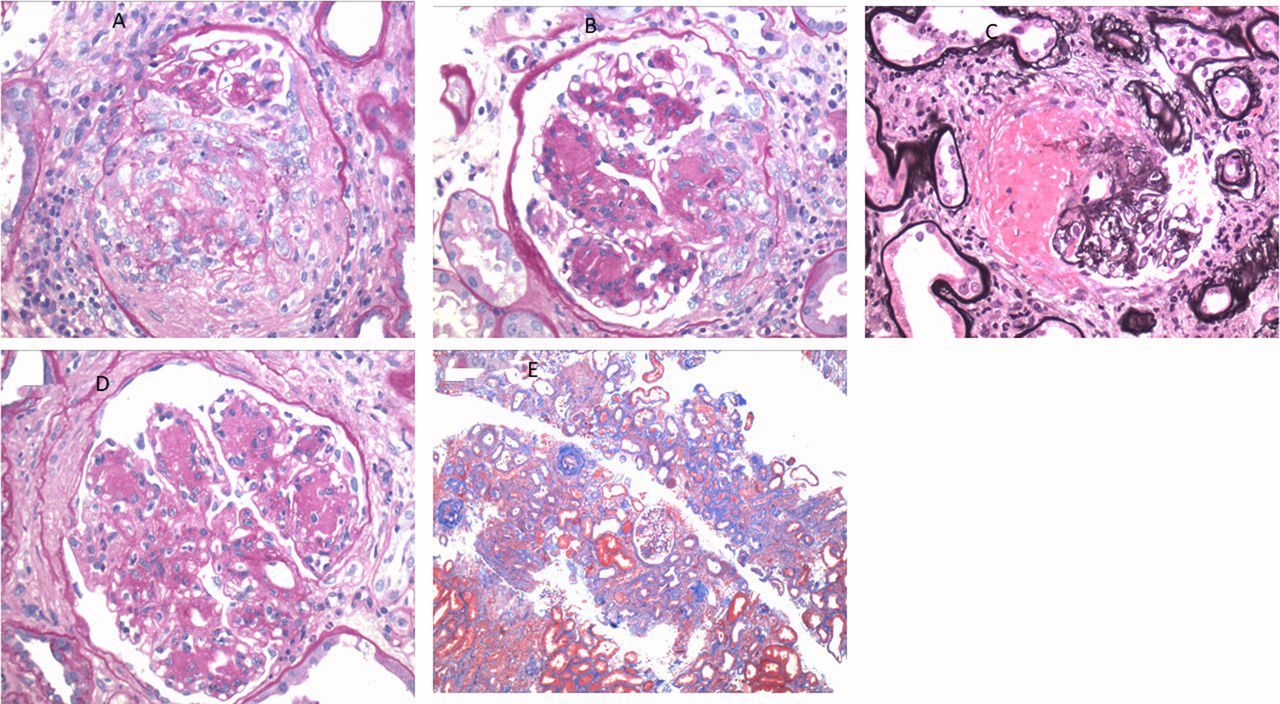{kind=link}
(A)Glomeruli with a fibrocellular crescent (H&E; original magnification ×400). (B) Glomeruli with a cellular crescent (H&E; original magnification ×400). (C) Glomeruli with segmental fibrinoid necrosis (methenamine-silver stain; original Magnification x400). (D) Glomeruli with nodular sclerosis (H&E; original magnification ×400). (E) Moderate tubular atrophy interstitial fibrosis involving 50% of the cortical area with moderate interstitial inflammation (Masson’s trichrome stain; original Magnification x200).
Diagnosis
Focal necrotising and crescentic GN, acute and chronic, with low-level immune deposits, superimposed on moderate to severe nodular diabetic glomerulosclerosis with moderate to severe tubular atrophy and interstitial fibrosis.
Clinical follow-up
The patient was treated with three doses of 1 g pulse intravenous methylprednisolone followed by oral prednisone 60 mg daily. She was started on rituximab infusions weekly (375 mg/m2/week) for 4 weeks. She required regular haemodialysis 5 days into her hospitalisation. On day 10 of her hospitalisation, she went into acute hypoxic respiratory failure with a non-contrast CT scan showing diffuse and extensive bilateral lung parenchymal infiltrates throughout lung fields with lung volume loss and pleural fluid. A bronchoscopy showed diffuse alveolar haemorrhage. She further treated with single volume exchange therapeutic plasmapheresis with fresh frozen plasma for seven sessions.
Outcome and follow-up
At the time of writing, the patient remained clinically stable but dependent on haemodialysis therapy for more than 12 months.
Discussion
AAV is a well-described clinical entity with an incidence of 10–20 cases per million,4 though very little is known regarding the prevalence of drug-induced vasculitis. Literature is available relating medications to vasculitis as early as the 1940s.2 This theory was supported after the discovery of ANCAs and their target antigens PR3 and MPO in the 1980s, with case series of patients who were ANCA positive and exposed to medications, such as, hydralazine, allopurinol and propylthiouracil, some developed vasculitis.5 6 Exposure to such drugs can provoke an immune reaction that results in the autoantibodies generation and clinical autoimmune disease, including immune complex or pauci-immune GN.2 Evidence of hydralazine-associated vasculitis including rapidly progressive GN dates to the pre-ANCA era.2 Hydralazine-induced vasculitis has an incidence of 5.4% in patients on 100 mg/day to 10.4% with 200 mg/day for >3-years duration, predominantly in patients who are slow acetylators.3 Choi et al7 performed a retrospective examination of drug-associated AAV in 250 MPO-positive AAV patients. In 30 patients with the maximum anti-MPO antibody titers, 10 patients exposed to hydralazine, of whom nine had renal involvement and five showing renal biopsy-proven pauci-immune necrotizing GN. Yokogawa and Vivino,8 in 2009, found 68 hydralazine vasculitis reports. The subjects in the study were predominantly female and had a mean duration of drug exposure of 4.7 years; with a mean dose of 142 mg/day. Renal disease was common on presentation (81%), and patients had further serological evidence of an autoimmune process (96% ANA positive, 26% anti-dsDNA antibody positive and 44% hypocomplementemia), similar to the previous study by Choi et al7. Kumar et al,9 studied 323 cases of AAV of which 12 exposed to hydralazine. Eight out of 12 patients presented with pulmonary infiltrates and one with haemoptysis. Only six patients had a renal biopsy and all of them had pauci-immune crescentic GN. The mean duration of therapy was 22 months with a mean cumulative dose of 146 g. Median age was 70.3 years with all patients being Caucasian with 58.3% female patients. All 12 patients in the study had elevated anti-MPO antibody.
The identified risk factors that predispose to hydralazine-induced AAV include a cumulative dose of more than 100 g, female gender and thyroid disease.10 Other risk factors identified include the human leucocyte antigen (HLA)-DR4 genotype, slow hepatic acetylation and the null gene for C4.11
We performed a literature search and in table 1, we reviewed the hydralazine-associated AAV with pulmonary–renal syndrome patients, with only 24 cases reported so far. Seventeen out of 24 patients in the case report survived. Our patient survived, though dependent on the dialysis 12 months after the initial presentation.
Hydralazine induced antinuclear cytoplasmic antibody vasculitis-patients with the pulmonary–renal syndrome, treatment and outcomes.
The patient in our case had diabetes for more than 30 years. To date, there have been only 10 reported cases of diabetic nephropathy with antineutrophil cytoplasmic antibodies mediated nephritis.12 Of the 10 cases, only three cases showed the pathological evidence of diabetic nephropathy. Approximately 20%–30% of diabetic nephropathy cases also complicated by nondiabetic nephropathy.13 Thus, our case also highlights the need for a higher degree of clinical suspicion in patients with worsening of renal function or abnormal urinalysis that does not concur with the natural course of diabetic nephropathy.
The mechanisms of hydralazine-associated AAV pathogenesis is not clear. In vivo study data suggests that ANCAs are by themselves pathogenic.14 MPO knockout mice that lack functioning B- and T-lymphocytes, when injected with anti-MPO splenocytes, developed severe necrotizing crescentic GN and haemorrhagic pulmonary capillaritis. It’s hypothesised that hydralazine collects in neutrophils and binds to MPO, this induces neutrophil apoptosis and cytotoxic products which further act as a source of immunogens as apparent by the presence of numerous antibodies that associated with hydralazine-induced AAV.15 The antibodies either alone or by a complex interaction with infectious agents or genetic factors may contribute to the disease. A study done by Magro et al suggested that hydralazine interferes with neutrophil extracellular traps (NETs), which leads to further uptake of previously sequestered antigens.16 Antibodies associated with hydralazine-induced vasculitis include MPO-ANCA, ANA, antihistone antibody, antielastase antibody and antiphospholipid antibody.15 17 Antihistone antibody is commonly seen with drug-induced vasculitis and is absent with ANCA-associated vasculitis.14 The combination of antihistone antibody, MPO and/or PR3 ANCA and absence of anti dsDNA antibody could be used to support the diagnosis of hydralazine-induced vasculitis in the appropriate clinical setting with evidence of pauci-immune GN.18 As the presence of multi-antigenicity is a key finding, multiple serological markers should raise suspicion for drug-induced vasculitis.19 Presence of very high MPO antibody titers is another distinct characteristic of drug-induced pauci-immune GN.7
The infrequency of drug-induced AAV and overlying features with idiopathic vasculitis is the greater dilemma in early diagnosis.19 The diagnosis relies mainly on constitutional and system-specific symptoms, exhaustive medication history, the total duration of drug therapy, the presence of serological markers and the resolution of symptoms after discontinuation of the offending drug.19 Patients with hydralazine-associated vasculitis characteristically have a more severe course, predominantly due to renal vasculitis and therefore require more aggressive treatment.8 Concomitant occurrence of pulmonary haemorrhage is the most powerful predictor of death,20 21 necessitating early diagnosis and prompt initiation of treatment. For a definitive diagnosis, a renal biopsy is strongly encouraged which also ascertains the severity of disease and aids in prognostication. Apart from a detailed history, laboratory and pathological findings, the Naranjo adverse drug reaction probability scale was used in our case, which specified that the association of hydralazine to pulmonary–renal syndrome was probable.22
Currently, no guidelines are available and no randomised controlled trials conducted in the treatment of hydralazine and other drug associated AAV. Treatment should individualise to the patient, based on the age, disease severity, co-morbidities and renal function on presentation. In mild cases, discontinuing the offending medication may lead to resolution of AAV. In severe cases, particularly with pulmonary or renal involvement, aggressive management with immunosuppressive regimens are needed for hydralazine and drug-mediated AAV. Corticosteroids with cyclophosphamide or rituximab and therapeutic plasma exchange for the pulmonary haemorrhage and rapidly progressive GN should consider. Immunosuppression should be individualised, preferable with a regimen with fewer side effects and a shorter duration of treatment. It is important to educate the patients about the future drug exposure, with the index drug added to the allergies list in the patient’s medical record. Further studies to better understand the etiopathogenesis of hydralazine-AAV is warranted to invent better markers for diagnosis and treatment.
Learning points
Hydralazine-induced antinuclear cytoplasmic antibody vasculitis and pulmonary–renal syndrome though rare, can be rapidly progressive and fatal.
A thorough clinical and medication history, serologies and pulmonary–renal pathological findings can help in the diagnosis.
Treatment includes stopping the hydralazine, appropriate use of immunosuppressants based on the severity and therapeutic plasmapheresis.
Acknowledgments
We would like to thank Dr Dominick Santorielli, MD, Assistant Professor of Pathology and Cell Biology, Columbia University, New York, for the renal biopsy pathology and images.
References
Footnotes
Contributors NRA, SP, AA and PJJ involved in the planning, conception, acquisition of data and helped with the final drafting of the case report. SP and NRA are the first authors and have equally contributed to the majority of the article.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Patient consent Not required.
Provenance and peer review Not commissioned; externally peer reviewed.