Article Text

Statistics from Altmetric.com
Description
A currently 12-year-old boy presented at the age of 8 months with sleepiness, irritability and a tense fontanelle with upgaze palsy on a background of a 2-month history of vomiting, difficulty feeding and weight loss. Acute communicating hydrocephalus was diagnosed using ultrasonography and CT and treated with Ventriculo-Peritoneal shunt insertion. Further investigation with MRI (2006) found a thoracic intramedullary mass with further intracranial and spinal leptomeningeal dissemination (LD) (figures 1 and 2). A biopsy of the thoracic intramedullary mass showed pilocytic astrocytoma (PA) (figure 3).

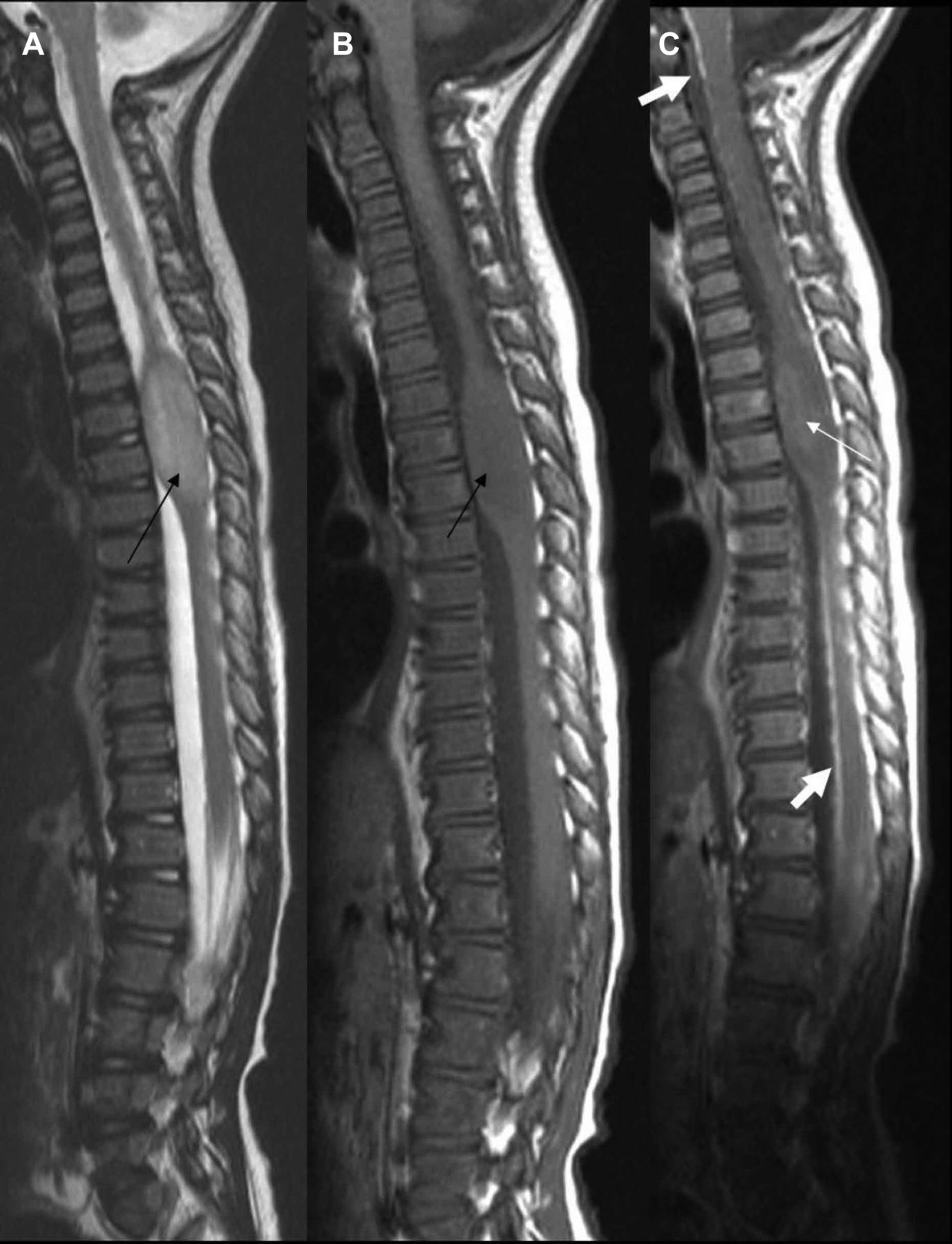
Initial MRI whole spine (2006). (A) T2 sagittal, (B) T1 sagittal and (C) postcontrast T1 sagittal. Black arrows in (A) and (B) show upper thoracic intramedullary mass. The white arrow in (C) shows no significant enhancement. Thick white arrows in (C) show leptomeningeal enhancement.

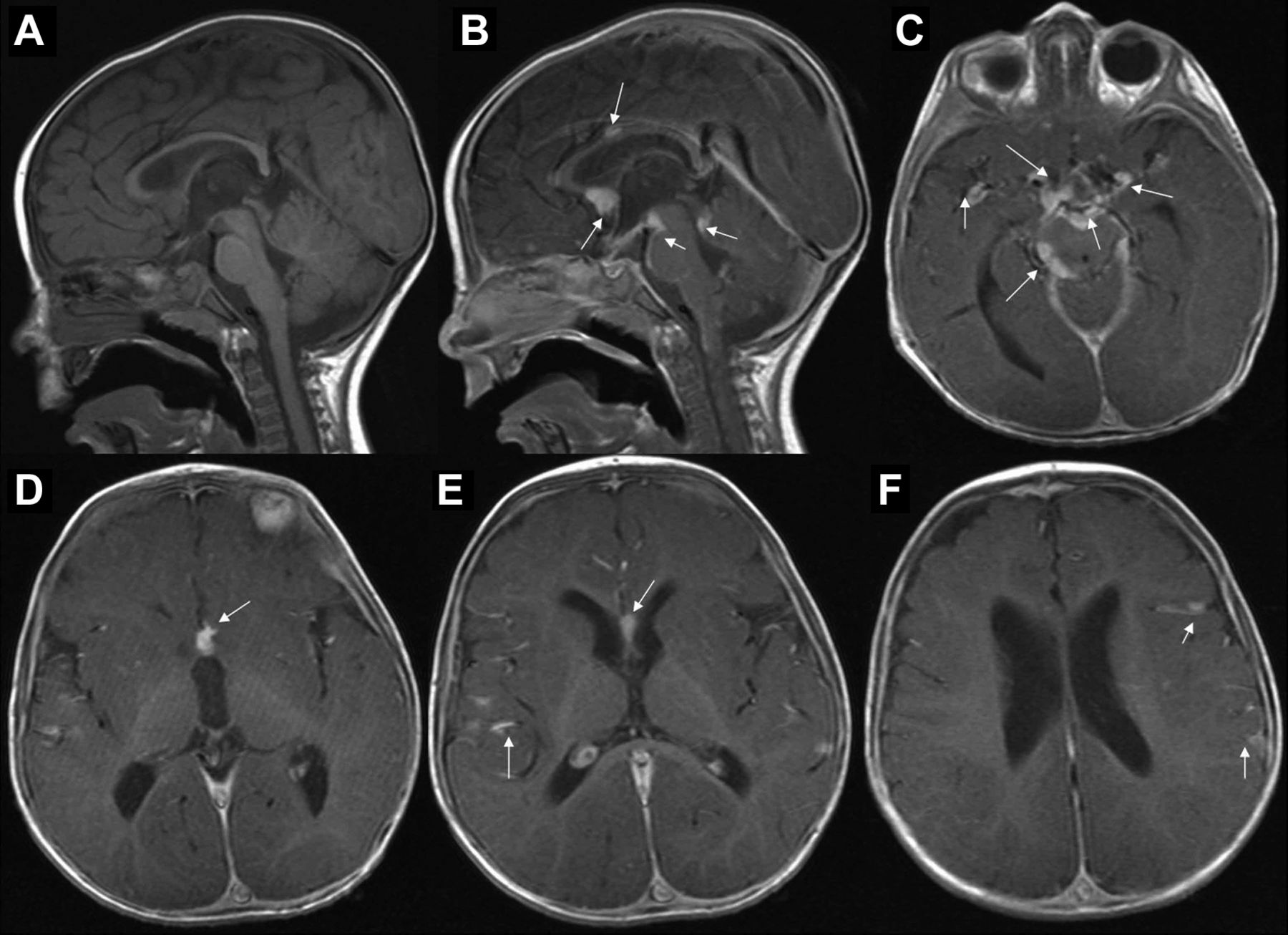
Initial MRI brain (2006). (A) T1 sagittal without contrast, (B) T1 sagittal with contrast, (C–F) T1 axial postcontrast images. White arrows in (B–F) show several areas of leptomeningeal dissemination.

Histopathology shows solid and less solid areas with Rosenthal fibres and elongated cell nuclei with long fibrillary processes consistent with pilocytic astrocytoma.
He received intensive 85 weeks of chemotherapy (2006–2008) with low-grade glioma protocol (SIOP-LGG 2004), including Vincristine (1.125–1.5 mg/m2), carboplatin (412–550 mg/m2) and etoposide (100 mg/m2). He responded well with almost complete resolution of metastatic disease (figure 4). There was no progression for several years until December 2016, when he developed a suprasellar/prepontine cystic area/loculated fluid with small eccentric enhancing nodule (figure 5). On further follow-up, the cyst has become more well defined in 2017 and 2018 scans with slight prominence of nodule (figure 6). The nodular enhancement in the spinal intramedullary lesion has also become slightly more prominent since June 2017 (figure 6) with otherwise overall stable appearances. There has been however no change in the neurological status. He is currently neurologically and developmentally well. His main problem has been progressive thoracic and lumbar scoliosis secondary to thoracic tumour; stabilised with bracing since 2011. Due to concerns of destabilisation, and following pubertal maturation, the patient underwent elective posterior spinal fusion T2–4 with instrumentation and bone graft. He underwent posterior spinal fusion in 2017. He remains well, 10 years post-treatment, with ongoing disease surveillance.

Postchemotherapy scan (2008). (A) T2 sagittal of spine. (B) T1 postcontrast sagittal of spine. (C,D) T1 postcontrast axials brain. (A,B) show stable appearances of intramedullary mass and resolution of leptomeningeal dissemination. (C,D) show resolution of intracranial leptomeningeal dissemination.

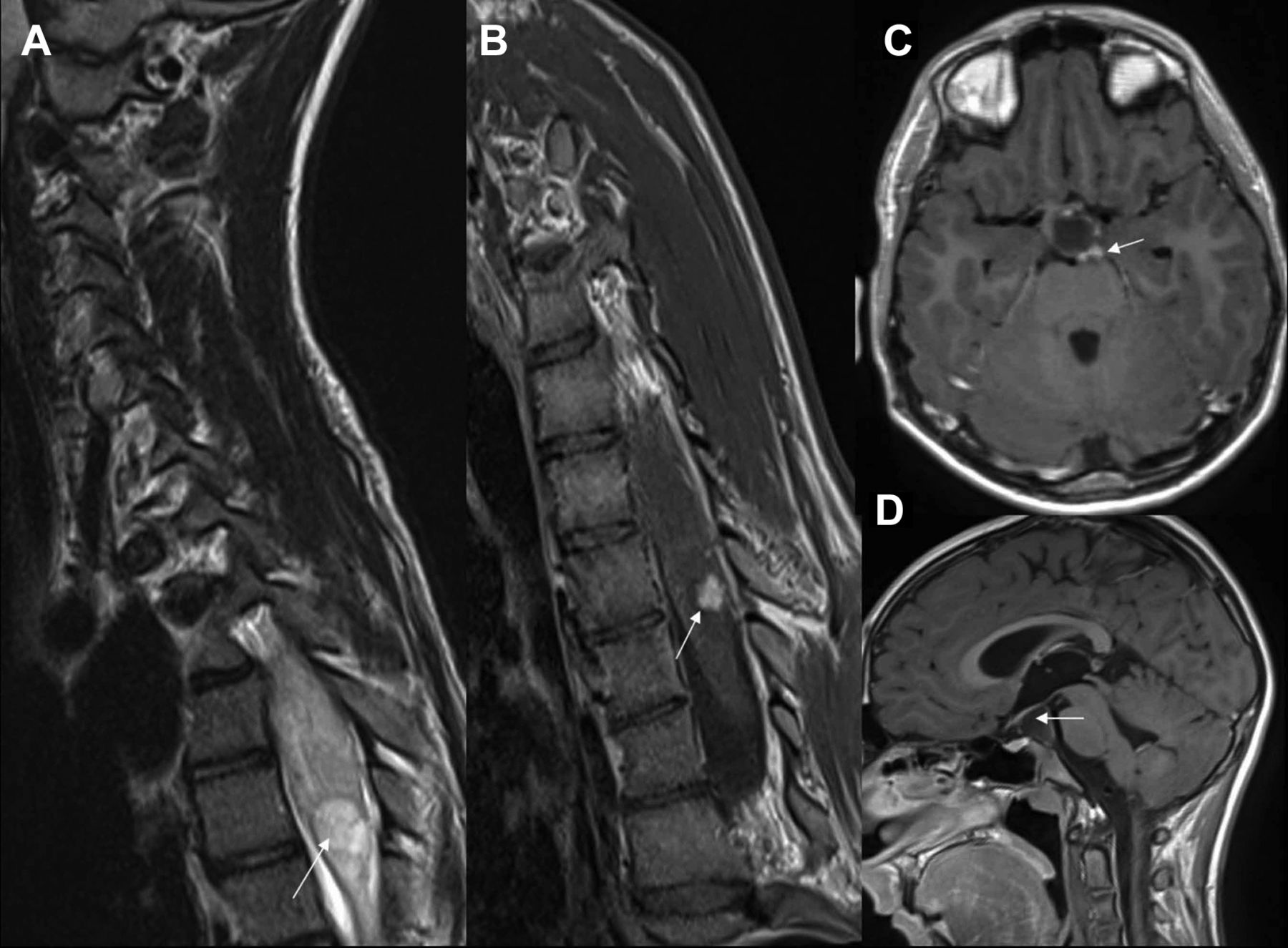
Follow-up scan (2016). (A,B) T2 and T1 sagittal postcontrast images of spine. (C–E) T1 postcontrast axial, sagittal and coronal images of brain. (A,B) show stable appearance of intramedullary mass (white arrows). Thick white arrow in (D) shows a cerebrospinal fluid/fluid loculation in suprasellar region with draping of optic tract over it (broken arrow). Thin white arrows in (C,E) show small eccentric enhancing nodule on the left side.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Follow-up scan (2017). (A,B) T2 and T1 sagittal postcontrast images of spine. (C,D) T1 postcontrast axial and sagittal images of brain. (A,B) show increasing scoliosis. White arrow in (B) show nodular enhancing lesion slightly more prominent than previous images. White arrow in (C) shows nodular enhancement in suprasellar region. White arrow in (D) shows more well-defined fluid/loculated component.
PA is the most common paediatric brain tumour accounting for ~20% of paediatric brain tumours.1 PA usually arise in the posterior fossa, hypothalamic region or optic chiasm (often associated with neurofibromatosis-1), occasionally in the cerebral hemispheres, and extremely rarely, in the spinal cord. Almost all are associated with single abnormalities of the mitogen-activating-protein-kinase pathway. While PA are grade 1 tumours, they occasionally can exhibit more aggressive behaviour such as malignant transformation, recurrence or LD.2 3 LD in PA is a rare occurrence, with less than 100 reported cases in the literature and even more rarely occurring with a spinal primary with only four reported cases.2 4 Around half of cases of LD present at diagnosis.1 It is not known, why LD, particularly with a spinal primary is such a rare occurrence. There is a possibility that it represents a distinct entity or has a specific phenotype; however, this will need further analysis. While LD can present with hydrocephalus, many are identified incidentally.4 Since the number of known LD are very low, there are no known factors described to suggest its occurrence; however, any change in the neurological status and any clinical suspicion of raised intracranial pressure should prompt further imaging to exclude its occurrence. PA is primarily treated with surgery, and occasionally, with radiotherapy and/or chemotherapy, while targeted therapies hold promise. Prognosis is excellent; with 10-year survival >90%, falling to <50% for patients with LD.4 As this case demonstrates however that despite LD, these patients can still have a relatively long and indolent course. This case report is intended to serve as a reminder of this rare and often chronic condition.
Learning points
Pilocytic astrocytomas, while grade 1 tumours, can rarely metastasise with leptomeningeal dissemination.
Leptomeningeal dissemination can present incidentally, either at diagnosis or later, but can be symptomatic and may present as hydrocephalus.
Despite early leptomeningeal dissemination, the patients can have a long and indolent course with a relatively stable disease over long periods.
Acknowledgments
We thank Dr Antonia Torgersen, Consultant Neuropathologist, for providing pathology image.
Footnotes
Contributors All authors (EB, AKK, BG, AE) have contributed substantially to the concept and manuscript including planning, conduct, conception and design, acquisition of data, analysis and interpretation of data.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Patient consent Parental/guardian consent obtained.
Provenance and peer review Not commissioned; externally peer reviewed.