Article Text

Summary
We experienced a 6-year-old case of drug-induced hypersensitivity syndrome (DiHS)/drug reaction with eosinophilia and systemic symptoms (DRESS) with subsequent development autoimmune thyroiditis (Hashimoto’s thyroiditis), type 1 diabetes with antithyroglobulin, thyroid peroxidase, insulinoma-associated antigen and anti-insulin antibodies at 4 months, alopecia at 7 months, vitiligo, uveitis due to Vogt-Koyanagi-Harada disease at 8 months after clinical resolution of the DiHS/DRESS. He was diagnosed as type III polyglandular autoimmune syndrome (PASIII) after DiHS/DRESS. Prompted by this case, we sought to determine which triggering factors were responsible for later development of PASIII in previously published cases with autoimmune sequelae. In the literature review, five patients with DIHS/DRESS were found to develop autoimmune sequelae consistent with PASIII. All cases with PASIII were much younger than those without them. Four out of the five patients were treated with intravenous immunoglobulin or pulsed prednisolone in the acute stage, although effective in short-term outcomes.
- dermatology
- therapeutic indications
- skin
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
Type III polyglandular autoimmune syndrome (PASIII) is a disorder characterised by the coexistence of more than two glandular autoimmune diseases, such as type 1 diabetes.1 Little information is available on critical triggers in the pathogenesis of PASIII in genetically predisposed individuals. Of note, very frequent component autoimmune diseases of PAS III reportedly occur as sequelae of a distinct type of severe drug eruption, drug-induced hypersensitivity syndrome (DiHS)/drug reaction with eosinophilia and systemic symptoms (DRESS); this syndrome is characterised by sequential reactivations of herpes viruses due to functional defects in regulatory T cells (Tregs).2–5 Paediatric DiHS/DRESS is often misdiagnosed as Kawasaki disease due to similar clinical presentations and treated with intravenous immunoglobulin (IVIG).
We describe a child case of DiHS/DRESS with subsequent development of PASIII. In this case, IVIG used would have served to accelerate progression to PASIII, although beneficial in short-term outcomes. Surprisingly, all previously published DiHS/DRESS cases with autoimmune sequelae had clinical features consistent with PASIII and most had been treated with pulsed prednisolone and/or IVIG in the acute stage.
Case presentation
Case report
A 6-year-old boy presented with erythematous lesions on his trunk and extremities. Two months prior to this visit, he suffered a fracture of the right upper arm and received a surgical operation. After the operation, he developed osteomyelitis and was treated with sulfamethoxazole–trimethoprim. Forty-five days later, erythematous plaques began on his extremities and trunk. Sulfamethoxazole–trimethoprim was withdrawn, but his conditions became worse and the patient was referred for further diagnostic workup and treatment to our hospital.
On examination, the patient had numerous 2–5 cm purplish and erythematous plaques on the back and extremities (figure 1A). The patient was hospitalised with fever of 38°C. Physical examination revealed leucocytosis (14.5X109/L) with atypical lymphocytosis (2.9X109/L), but no eosinophilia (0.5X109/L), liver dysfunction (alanine aminotransferase (ALT) 357 U/L) and cervical lymphadenopathy (figure 2). A skin biopsy revealed a spongiotic change of the epidermis with scattered necrotic keratinocytes and a mild perivascular infiltrate. The diagnosis of DiHS/DRESS was made and oral prednisolone, 30 mg/day, was begun. The patient, however, once again experienced the similar erythematous plaques with a high-grade fever and a marked increase in liver enzyme levels (ALT 1857 U/L). On the 12th day of hospitalisation, he received IVIG, at a dose of 1g/kg/day for 2 days given his low immunoglobulin (Ig)G levels (386 mg/dL). After starting IVIG, he showed clearing of lesions and rapid improvement in liver enzyme levels. Because of frequent flaring of liver dysfunction and rashes, we decided to taper prednisolone more gradually over a prolonged period of time (5 mg/day per 2–3 week).
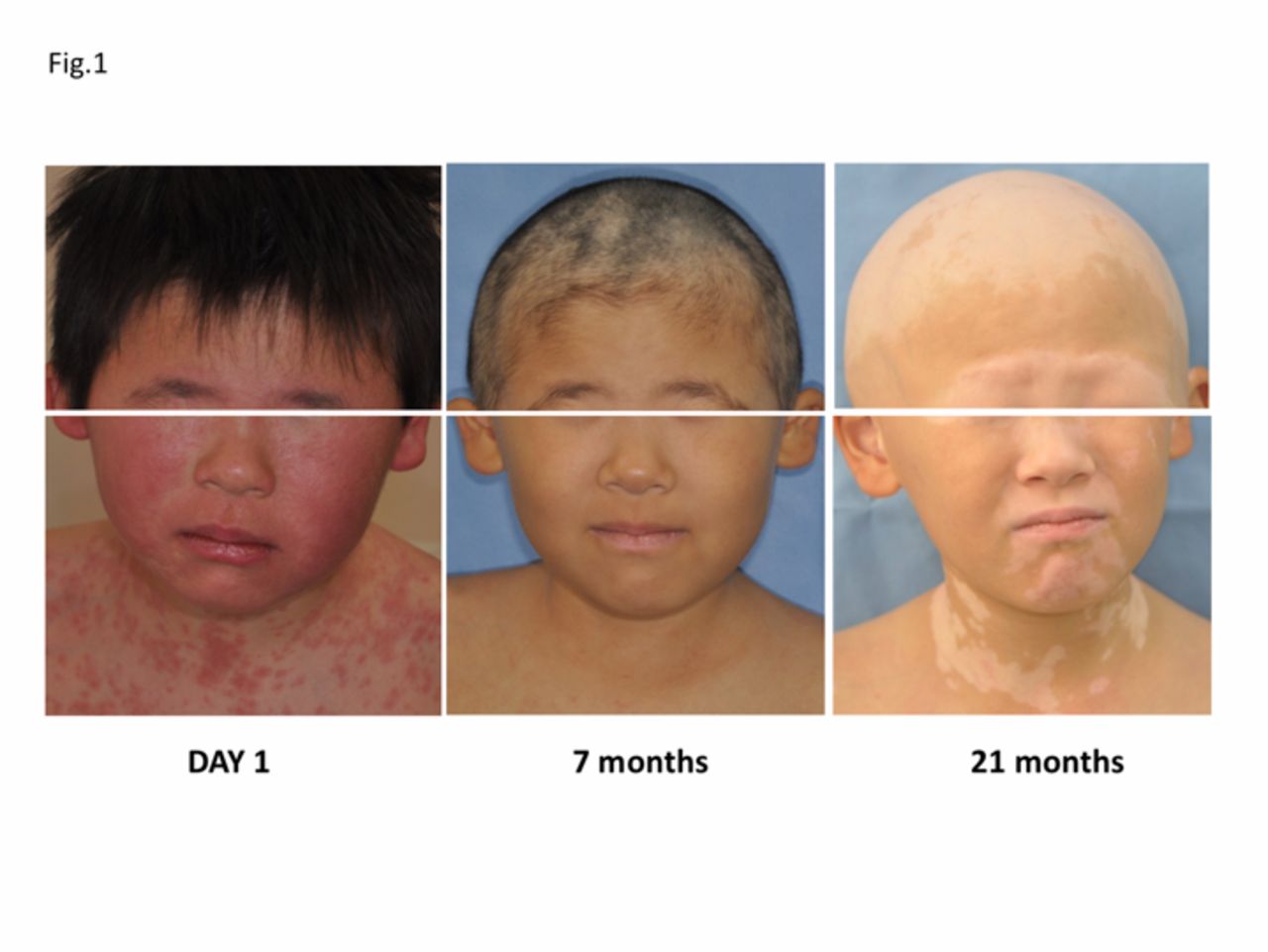
Alterations of clinical presentation of drug-induced hypersensitivity syndrome/drug reaction with eosinophilia and systemic symptoms and autoimmune sequelae. (A) At the initial presentation, the patient had purplish and erythematous plaques on the face. (B) The partial hair loss started 7 months after onset of the disease. (C) Total hair loss associated with generalised vitiligo after 21 months.

{kind=link}
{kind=link}
Clinical symptoms and laboratory findings in case 5 with type III polyglandular autoimmune syndrome (present case) after onset of drug-induced hypersensitivity syndrome/drug reaction with eosinophilia and systemic symptoms in relation to treatment. AST, aspartate aminotransferase; ALT, alanine aminotransferase; IVIG, intravenous immunoglobulin; PSL, prednisolone; WCC, white cell count.
After 4 months, the patient was again admitted to our hospital for nausea, fatigue and excessive thirst. Laboratory tests revealed a marked increase in fasting blood sugar, 580 mg/dL, haemoglobin A1c, 10.2%, anti-insulinoma-associated antigen 2 (IA-2) antibody titre 1.0 U/mL (normal <0.4 U/mL) and anti-insulin antibody titre 50.0 U/mL (<0.4 U/mL). He was diagnosed as having type 1 diabetes mellitus and treatment with insulin was initiated. Because laboratory tests also revealed an increase in antithyroglobulin antibody 36 IU/ml (<28 IU/mL) and antithyroid peroxidase antibody titre 113 IU/mL (<16 IU/mL), he was also diagnosed with Hashimoto’s disease. The doses of prednisolone were gradually reduced and eventually withdrawn on day 180.
On days 215–243, he developed alopecia and vitiligo on his back and upper extremities (figure 1B). On day 243, he complained of impaired vision in both eyes. Fundus examination showed multiple areas of neurosensory detachments and focal areas of multiple confluent yellow-coloured placoid choroidal lesions. A diagnosis of concurrent development of Vogt-Koyanagi-Harada disease and prednisolone was again increased to a dose of 5 mg/day. He developed alopecia totalis with generalised vitiligo 21 months after onset of disease (figure 1C).
Investigations
Prompted by this case, we investigated whether autoimmune diseases with various autoantibodies occurring in reported DiHS/DRESS cases could have clinical features consistent with PASIII. As shown in table 1, five published cases sequentially developing PASIII including this case, were found, although none have reported these autoimmune diseases in the name of PASIII.3 5–8 The mean age of these five cases (21±13.4 years) was much younger than that in our 55 institutional cases with DiHS/DRESS (54.5±2.7 years) (manuscript submitted): four of the five cases were categorised as paediatric (<18 years) DiHS/DRESS. Among the three subcategories of PASIII, the present case met the criteria for A and C type of PASIII.1 9
Characteristics of patients who developed PAS after drug-induced hypersensitivity syndrome/drug reaction with eosinophilia and systemic symptoms
Differential diagnosis
There are a number of differential diagnostic considerations in children; they include Kawasaki disease and viral eruptions, which show unexplained illness that includes rash, fever and lymphadenopathy. In particular, there is an emerging recognition that some children with DiHS/DRESS have been misdiagnosed as Kawasaki disease and erroneously treated by high doses of IVIG within the first 10 days of fever onset.
Treatment
Although clinical experience suggested that high doses of IVIG or pulsed corticosteroids were effective in abrogating the acute inflammation of DiHS/DRESS, our case and previously reported cases with PASIII suggested that these aggressive therapies in paediatric patients paradoxically increased the risk of future development of autoimmune sequelae, particularly PASIII. The potential reward should outweigh the potential risk, especially in a long-term outcome.
Outcome and follow-up
Most importantly, pulsed corticosteroids or IVIG was used for treatment of DiHS/DRESS during the acute stage in four of the five reported DiHS/DRESS cases who eventually developed PASIII, except for Case 1, in whom detailed information is not available. Although current literature recommends, DiHS/DRESS cases treated with these aggressive therapies in the acute stage, especially paediatric cases, developed PASIII several months after successful resolution of initial clinical symptoms.9–12
Discussion
Which triggering factors are responsible for later development of PASIII in genetically predisposed individuals after clinical resolution of DiHS/DRESS? Of note, the development of PASIII occurred after administration of a short course of pulsed prednisolone in three of the five reported cases (table 1). Some clues may come from studies of immune reconstitution inflammatory syndrome (IRIS), where a rapid immune recovery on withdrawal or reduction of immunosuppressive agents, paradoxically causes extensive pathology.13 Because a rapid and great reduction of prednisolone doses, for example, from 1 g/day to 60 mg/day over a period of 2 days, is necessarily needed in pulsed prednisolone therapy, thereby increasing the risk of IRIS, pulsed prednisolone therapy for DiHS/DRESS may serve to accelerate aggressive cytotoxic immune responses causing extensive pathology.
Although our patient’s young age is unusual in patients with DiHS/DRESS and PASIII, another unusual aspect was its low levels of IgG followed by a marked increase in the levels 1 month later, at which time its levels usually return to normal; this suggests that marked rebound of IgG synthesis may have occurred around the time of IVIG administration in this case: thus, the timing of IVIG administration was likely to coincident with the timing of the rebound and with the peak of liver dysfunction. In this regard, we previously demonstrated that both severe liver damage and defective Treg function in combination could be needed for the subsequent generation of autoantibodies in patients with DiHS/DRESS.14 In our patient, given the clinical course of the disease, IVIG would have been administered at or immediately after the peak of liver damage and rebound in IgG, A and M synthesis. This timing of IVIG administration may allow autoreactive B cells to function in an uncontrolled fashion: IVIG administration at the peak of severe liver damage could further accelerate the rapid recovery of B cells, thereby causing the subsequent expansions of autoreactive B cells with the specificity to a variety of epitopes. Supporting this hypothesis, De Grandmont et al observed that the addition of IVIG in culture of CD40L-stimulated B cells accelerated the differentiation of part of peripheral B cells into IgG-producing plasma cells in the presence of interleukin (IL)-4 and IL-10: importantly, secreted IgGs were shown to be reactive with antigens such as nucleoprotamine, dsDNA, tetanus toxin and human IgG F(ab’)2 fragments. The action of IVIG on DiHS/DRESS may be different depending on the state of activation of the B cells exposed to IVIG.15
In conclusion, paediatric patients with DiHS/DRESS exhibit an increased risk for subsequently developing PASIII. We recommend that patients with DiHS/DRESS, especially paediatric patients, be screened for the generation of various autoantibodies and that at-risk patients be advised to avoid therapies that can accelerate rapid recovery of B cells and T cells, such as IVIG and pulsed prednisolone in the acute stage.
Learning points
In literature review, five patients with drug-induced hypersensitivity syndrome (DiHS)/drug reaction with eosinophilia and systemic symptoms (DRESS) were found to develop autoimmune sequelae consistent with type III polyglandular autoimmune syndrome (PASIII) 2–7 (average 4.8) months after onset. Four out of the 5 patients were treated with intravenous immunoglobulin or pulsed prednisolone in the acute stage.
Paediatric or younger patients with DiHS/DRESS have the greatest risk for subsequently developing PASIII, when treated with intravenous immunoglobulin or pulsed prednisolone that can accelerate rapid recovery of B and T cells in the acute stage, although beneficial in the short-term outcome.
Footnotes
Contributors YA and TS contributed to the conception and design, interpretation of data and drafting the article or revising it critically for important intellectual content. All authors contributed to the acquisition of data or analysis. All authors contributed to the final approval of the version published; agreed to be accountable for the article and to ensure that all questions regarding the accuracy or integrity of the article are investigated and resolved.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Patient consent Guardian consent obtained.
Provenance and peer review Not commissioned; externally peer reviewed.