Article Text

Statistics from Altmetric.com
Description
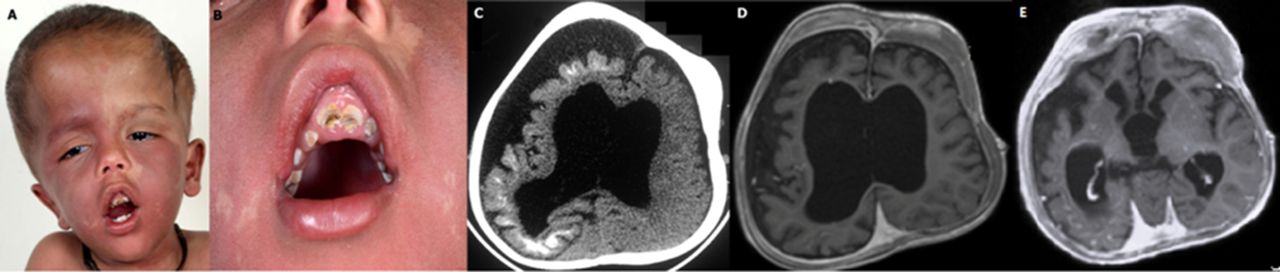
A 3-year-old boy presented with global developmental delay, abnormal craniofacial growth and left focal seizures since infancy. He was the first child of a non-consanguineous couple with unremarkable perinatal period. Family history was non-contributory. On examination, he had macrocephaly (head circumference 55 cm, >3 z-score), extensive port-wine stain distributed bilaterally over the face and the trunk, cloverleaf-shaped skull with facial distortion (figure 1A), marked gingival hyperplasia with dental malocclusion (figure 1B), bilateral blue sclera with tortuous engorged veins, brisk muscle stretch reflexes, left hemiparesis and bilateral Babinski’s sign. The rest of the systemic examination was unremarkable. A clinical diagnosis of angiodysplastic variant of Sturge Weber syndrome (SWS) with extensive bilateral facial and truncal port-wine stain, glaucoma and focal epilepsy was considered. CT of the brain revealed right-sided subcortical tram-track calcification and concordant parenchymal volume loss (figure 1C). MRI of the brain showed right-sided leptomeningeal angiomatosis with cerebral atrophy (figure 1D), thickened calvarium and fibrous dysplasia of the right frontal bone (figure 1D and E). He was treated with antiepileptic drugs (phenytoin 8 mg/kg/day and valproate 25 mg/kg/day) and aspirin (5 mg/kg/day). Multidisciplinary care involving follow-ups in ophthalmology, dentistry, neurosurgery, occupational therapy and physiotherapy was planned.
{kind=link}
(A–B) Clinical photograph of the patient showing the characteristic cloverleaf-shaped skull growth, bilateral port-wine stain, blue sclera, gingival hyperplasia and dental malocclusion. (C) Plain CT scan of the brain showing right-sided subcortical tram-track calcification and concordant parenchymal volume loss and dilatation of ventricles. (D) MRI of the brain showing right cerebral hemiatrophy with meningeal enhancement and gyral calcification suggestive of Sturge Weber syndrome and (E) fibrous dysplasia in the frontal bone and right subdural collection.
SWS is a sporadic, neurocutaneous syndrome defined by the association of a facial capillary malformation with ipsilateral glaucoma and vascular malformation of the brain (leptomeningeal angioma). Mild osseous hypertrophy of the calvarium ipsilateral to the port-wine stain in SWS is well known and is proposed to be either due to a compensatory response to underlying brain parenchymal atrophy or due to focal venous hypertension associated with primary venous dysplasia.1 However, massive osseous and soft tissue hypertrophy leading to facial distortion and compressive effects is unusual and has been termed angiodysplastic SWS.2 The underlying mechanism for this substantial osseous and soft tissue distortion remains conjectural. Proposed mechanisms include abnormal physiological alternation of haemodynamic and metabolic properties adjacent to or within the bone directly affecting bony growth; an intrinsic, congenital mesenchymal defect leading to growth alteration or secondary effects of the vascular malformation on the underlying bone predisposing to mutations and thus neoplastic transformations.3 Klippel-Trenaunay-Weber syndrome is an important clinical differential diagnosis for osteohypertrophy associated with angiomatosis but, unlike SWS, the former syndrome is characterised by cutaneous capillary malformation with underlying venous varicosity and strong predilection for hypertrophy of the extremities. Hence, a differentiation should be made between the two clinical entities and children with SWS should be referred early for expert consultation and management of specific problems. Children with angiodysplastic variant of SWS need regular follow-up to look for malignant transformation of dysplastic tissue. The mainstay of management is supportive and includes anticonvulsants for seizures; antiglaucoma measures; prophylactic aspirin therapy and neurorehabilitation. Surgical reduction of symptomatic osseous hypertrophy may be useful.
Learning points
Massive osseous and soft tissue hypertrophy leading to facial distortion and compressive effects is unusual in classic Sturge Weber syndrome (SWS).
Children with angiodysplastic variant of SWS need regular follow-up to look for malignant transformation of the dysplastic tissue.
Footnotes
Contributors AK and AGS: draft of the manuscript and revision. SV: draft of the manuscript and radiological review. PS: clinician in charge, plan of the study and final review of the manuscript.
Competing interests None declared.
Patient consent Parental/guardian consent obtained.
Provenance and peer review Not commissioned; externally peer reviewed.