Article Text

Statistics from Altmetric.com
Description
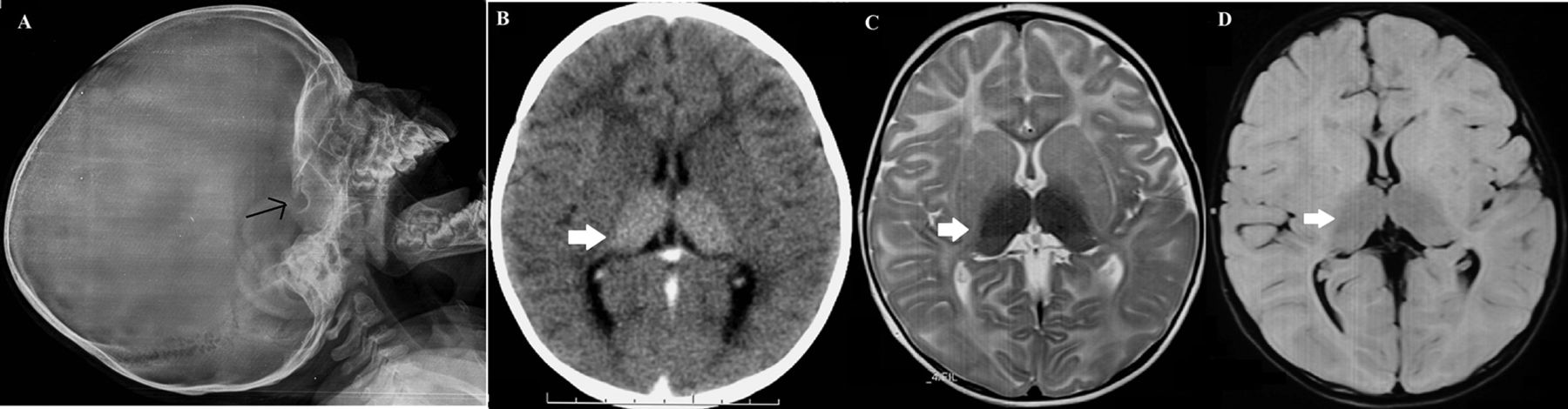
We evaluated a boy aged 16 months with developmental arrest at the age of 6 months followed by neuroregression and recurrent generalised seizures. Perinatal and family history was not contributory. He was first born to non-consanguineous parents by term, uncomplicated vaginal delivery and weighed 2.8 kg at birth. On examination, he was unable to hold neck, fixate, coo or smile and showed no interest in the surroundings. His weight was 9.5 kg (between 15th and 50th centile), length 78 cm (between 15th and 50th centile) and head circumference 44.4 cm (below 3rd centile) with normal head circumference of father (54 cm) and mother (51 cm). He also had hyperacusis, bilateral cherry-red spot, generalised hypotonia, brisk muscle stretch reflexes, bilateral Babinski’s sign and no organomegaly. In view of infantile-onset neuroregression, microcephaly, seizures, cherry-red spot and spasticity, clinical diagnoses of GM2 (Tay-Sach’s and Sandhoff’s disease), GM1 gangliosidosis and Krabbe’s disease were considered initially. Skull radiograph showed J-shaped sella turcica (figure 1A). CT scan of brain showed bilateral thalamic hyperdensity (figure 1B). MRI of the brain showed bilateral thalamic T2/fluid-attenuated inversion recovery sequence hypointensity with diffuse white matter hyperintensity (figure 1C-D). Based on the clinical and radiological findings, a diagnosis of an infantile-onset GM2 gangliosidosis was concluded. Enzyme analysis showed deficient total hexosaminidase (A+B) enzyme in plasma (<0.062 nmol/hour/mg, normal range 660–5000 nmol/hour/mg). Genetic counselling was offered to the parents. The patient succumbed to an intercurrent respiratory illness 2 months later. Sandhoff's disease is an autosomal-recessive lysosomal storage disorder due to hexosaminidase A and B enzyme deficiency (due to an abnormal β-subunit) leading to accumulation of glycosphingolipids in neuronal cells and subsequent neurodegeneration.1 Progressive systemic accumulation of sphingolipids leads to macrocephaly, cherry-red spots in the eye, skeletal dysostosis and organomegaly.2 It is unusual to see microcephaly in storage disorders and may cloud the clinical diagnosis. Anecdotal reports of microcephaly in 3 of the 18 Iranian patients with GM2 gangliosidosis support our finding.3 The index case presented with the classical features of bilateral cherry-red spot, skeletal storage and psychomotor arrest. In the absence of familial small head size or significant malnutrition, it is intriguing to see microcephaly in association with Sandhoff's disease. It may be attributed to the onset of significant neuronal loss in early infancy secondary to GM2 ganglioside deposition. Contrary to the classical teaching, our case highlights that the absence of macrocephaly or presence of microcephaly should not deter a clinician from suspecting GM2 gangliosidosis in the correct clinical context.

{kind=link}
(A) Lateral skull radiograph showing the J-shaped sella turcica (arrow). (B) Contrast-enhanced CT scan of the brain showing bilateral hyperdense thalami (arrow) and abnormally hypodense white matter in bilateral frontal area in axial section. MRI (C) T2-weighted and (D) FLAIR axial sections showing bilateral hypointense thalami (arrows) and hyperintense periventricular and subcortical white matter consistent with GM2 gangliosidoses.
Learning points
Infantile Sandhoff's disease may present with microcephaly.
Absence of macrocephaly should not deter a clinician to diagnose Sandhoff's disease in the appropriate clinical context.
Thalamic hypointensities on brain MRI are an important radiological clue.
Footnotes
Contributors KM: patient management, draft of manuscript. RSK: patient management, draft of manuscript. PS: analysis of radiological data, critical review of manuscript for intellectual content and final approval of the version to be published. AGS: patient management, critical review of manuscript for intellectual content and final approval of the version to be published.
Competing interests None declared.
Patient consent Obtained from guardian.
Provenance and peer review Not commissioned; externally peer reviewed.