Article Text

Summary
The overall incidence of neurological manifestations is relatively low among patients with mixed connective tissue disease (MCTD). We recently encountered a case of autoimmune adhesive arachnoiditis in a young woman with 7 years history of MCTD who presented with severe back pain and myeloradiculopathic symptoms of lower limbs. To the best of our knowledge, adhesive arachnoiditis in an MCTD patient has never been previously reported. We report here this rare case, with the clinical picture and supportive ancillary data, including serology, cerebral spinal fluid analysis, electrophysiological evaluation and spinal neuroimaging, that is, MRI and CT (CT scan) of thoracic and lumbar spine. Her neurological deficit improved after augmenting her immunosuppressant therapy. Our case suggests that adhesive arachnoiditis can contribute to significant neurological deficits in MCTD and therefore requires ongoing surveillance.
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Background
Mixed connective tissue disease (MCTD) is a well-defined entity with a wide spectrum of clinical manifestations. Some patients initially diagnosed with MCTD eventually manifest symptoms more consistent with systemic lupus erythematosus (SLE) and vice versa.1
Neurological manifestations are reported in 10% cases of MCTD.2 Recent studies suggest that prevalence may be greater than reported before,3 ,4 involving central and peripheral nervous system. The most common disorders are trigeminal neuralgia, vascular-type headache, aseptic meningitis, psychosis and convulsions.4 ,5 Isolated cases of intracranial haemorrhage, cauda equina syndrome, transverse myelitis, optic neuropathy and retinal vasculitis have been reported as well.6–10
Adhesive arachnoiditis is a relatively uncommon chronic pathological disorder, characterised by an inflammatory insult to the arachnoid layer of the meninges that leads to fibrosis. As a sequel, the arachnoid becomes abnormally thick and adherent to the surrounding layers of pia and dura mater. The subsequent abnormal adhesion of nerve roots to the dural sac or to each other (clumping) can produce neurological impairment. The usual symptoms of arachnoiditis are severe back pain, paraesthesia, lower limb weakness and dissociative sensory loss. Common causes are prior spinal surgery, spinal inflammation or infection such as tuberculosis meningitis, trauma, haemorrhage, injection of anaesthetic agents and oil-based myelographic contrast agents. It is diagnosed on clinical grounds and supportive MRI findings.11 ,12
The pathogenesis of adhesive archnoiditis has not been fully elucidated. We present to the best of our knowledge the first case of adhesive arachnoiditis in an MCTD patient that resulted in myeloradiculopathic symptoms leading to significant neurological comprise. This manuscript also captures the challenges of correct diagnosis and subsequent management of this uncommon debilitating clinical entity.
Case presentation
A woman aged 33 years presented with 2-year history of low back pain, getting worse within last couple of months.
Seven years ago, she was diagnosed as MCTD when she presented to rheumatology service with polyarthritis, photosensitive butterfly rash, fatigue and scarring alopecia. Laboratory investigations showed raised ESR (25 mm/hour, N=5–12), normal C reactive protein, polyclonal hypergammaglobulinemia (IgG=23.74 g/L, N=5.4–16.1: IgA=5.11 g/L, N=0.9–3.4), C4 hypocomplementemia (0.11 g/L, N=0.16–0.38), positive serology for ANA (titre1/400,speckled pattern), anti-U1RNP and anti-Ro antibody and negative for anti-dsDNA antibody and antineutrophil cytoplasmic antibody. Her urine examination was normal.
She was a non-smoker, non-alcoholic, professional beauty therapist who was happily married and had two successful pregnancies with full term normal delivery. There was no history of rheumatological diseases in her family.
She was treated with immunosuppressants (hydroxy chloroquine/azathioprine/mycophenolate mofetil), oral steroids and aspirin over the course of her disease. She had a number of acute exacerbations, requiring steroids, typified by fatigue, hair loss and arthralgia. There had been no major systemic aspects of MCTD in conjunction with the neurological symptoms in last few years.
On her recent presentation, she reported of severe lower back pain radiating to right leg associated with pins and needles from waist down, 2–3 episodes of faecal incontinence, poor balance, perineal and perianal numbness and globally altered sensations in both legs. She acknowledged involuntary jerking of both lower extremities at night. Clinically, she had restricted right straight leg raise test, absent right knee jerk and diminished bilateral ankle jerks. Sensory examination showed diminished pinprick sensation in both extremities, more pronounced on right, extending up into the waist in a symmetrical distribution to the T10 level with no sacral sparing. Her Romberg's test was positive. There was no abnormality in cranial nerves, upper extremities or upper trunk. Rest of deep tendon reflexes were well preserved with good 2-point discrimination, vibration, proprioception, pinprick and temperature sensation. Muscle bulk and tone was also preserved in upper and lower limbs with flexor planter response.
Investigations
Laboratory investigations showed raised ESR (46 mm/hour, N=5–12 mm/hour), persistent C4 hypocomplementemia, positive ANA (titre1/1600, speckled pattern) and anti-U1RNP. This time her serology was positive for anti-dsDNA antibody (quantitative enzyme immunoassay=268, N=0–100), antiribosomal P-protein and negative for anticardiolipin IgG and anti-β-2 glycoprotein IgG antibody. Full blood count, renal, liver and bone profile was normal. At this stage, she clearly had MTCD with Lupus overlap.
On her urgent MRI lumbar-spine, conus medullaris and cauda equina were found within normal limits without any spinal cord signal abnormality, oedema or tumour. Her immunosuppressants (azathioprine and prednisolone) were up-titrated and given the huge impact of her symptoms limiting her activities of daily living, she received two fluoroscopic-guided caudal epidural injections 1 month apart with partial improvement in backache. Given the unusual nature of her symptoms, she underwent lumbar puncture and the cerebral spinal fluid (CSF) analysis was normal for white cell count (white cells <5), glucose and proteins. She underwent right lower limb neurophysiological studies (EMG/NCS) which did not report evidence of right L4 through S1 radiculopathy or sensorimotor polyneuropathy.
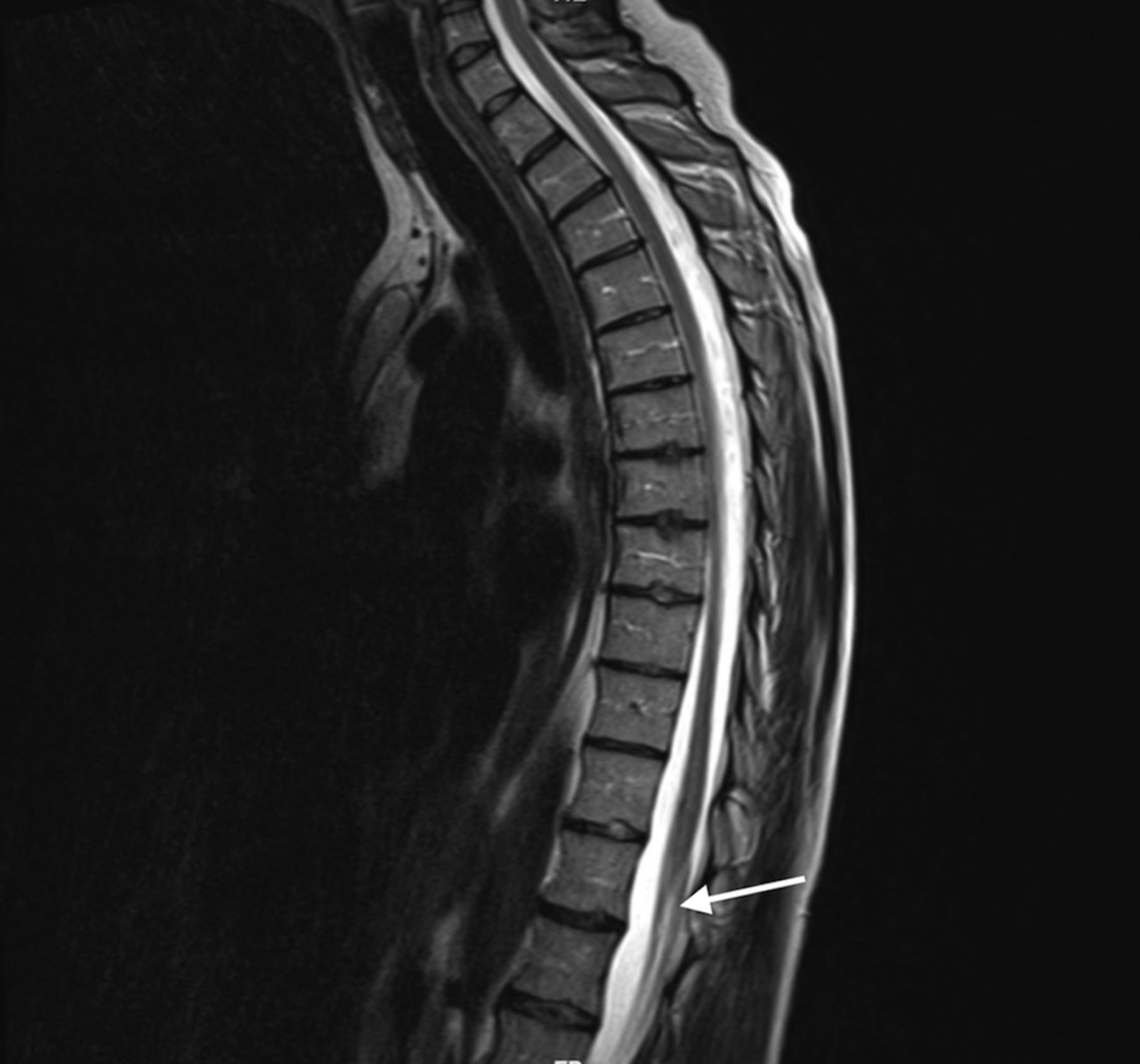
On account of worsening symptoms, she had a non-contrast CT scan of thoracolumbar spine, which documented thoracic spinal cord arachnoid calcification (figure 1) and raised concern for chronic adhesive arachnoiditis. It was further evaluated with MRI thoracolumbar spine which reported abnormally distributed lower lumbar spine and thecal sac nerve roots demonstrating clumping (figures 2 and 3) and augmented the existing concern for chronic arachnoiditis.

CT scan of thoracic spine: axial soft tissue window image (left) and sagittal reformatted image (right) demonstrating tram-like intrathecal calcifications (arrows) delineating the spinal cord compatible with arachnoiditis ossificans. Stars represent incidental finding of Schmorl's nodes.

Sagittal T2-weighted lumbar spine MRI demonstrating T2 hyperintense signal changes (arrow) consistent with thickened, clumped nerve roots.

{kind=link}
{kind=link}
{kind=link}
MRI lumbar spine: axial T1 postintravenous contrast image with fat suppression. The arrow points to the peripheral clumping of lumbar spinal nerve roots in the thecal sac.
Differential diagnosis
Our top differential diagnosis was cauda equina syndrome. Other possible differentials that could mimic symptoms of our patient were spinal cord tumours, syringomyelia, complex regional pain syndrome and multiple sclerosis which were ruled out by appropriate investigations. Failed back surgery syndrome is an important differential of adhesive arachnoiditis in postoperative phase.
Treatment
Our patient's imaging studies were discussed with neuroradiology and in the light of her background history of MCTD, recent unusual neurological presentation and results of investigations, especially neuroimaging findings, it was decided that she likely had adhesive arachnoiditis related to her connective tissue disorder (MCTD Lupus overlap) which possibly started at the time when she initially presented with backache and had progressed since then possibly due to scarring.
While she was on azathioprine for immunomodulation, rituximab (anti-CD20 monoclonal antibody) was added aimed at her neurological symptoms which successfully stopped progression of her symptoms.
Outcome and follow-up
Eighteen-month follow-up showed progressive improvement in her neurological symptoms with no recurrence. Her backache responded to analgesics.
Discussion
In 1972, Sharp et al13 described MCTD, an apparently distinct overlap syndrome sharing many features of SLE, scleroderma and polymyositis. As mentioned earlier, neurological manifestations are reported in 10% cases of MCTD.2 Recent studies suggest that prevalence may be greater than reported before,3 ,4 involving central and peripheral nervous system. The most common disorders are trigeminal neuralgia, vascular-type headache, aseptic meningitis, psychosis and convulsions.4 ,5 Isolated cases of intracranial haemorrhage, cauda equina syndrome, transverse myelitis, optic neuropathy and retinal vasculitis have been reported as well.6–10
Arachnoiditis, first described by Victor Horsley in 1909,14 is a rare inflammatory condition characterised by thickening of the arachnoid membrane and adhesions of dura mater that causes intractable lower back pain and various other devastating neurological complications that include cranial neuropathies, myelopathies and radiculopathies. The most common aetiological factors in the development of spinal arachnoiditis are infection, intrathecal injection of steroids or anaesthetic agents, trauma, subarachnoid haemorrhage, ionic myelographic contrast materials, multiple back surgeries and lumbar puncture.15 ,16 Adhesive arachnoiditis, the most severe type of chronic arachnoiditis, results in scar tissue formation, which compresses nerve roots and disrupts their blood supply and also normal flow of CSF. It can progress to arachnoiditis ossificans, an end-stage complication of adhesive arachnoiditis characterised by the pathological ossification of the spinal arachnoid.17 Arachnoiditis can also mimic the symptoms of other diseases, such as spinal cord tumours, cauda equina syndrome, arachnoiditis ossificans and syringomyelia.18 MRI is the gold standard in the diagnosis of arachnoiditis; however, unenhanced CT (CT scan) better elucidates the presence and extent of arachnoid ossifications and is thus interrelated to MRI.
The pathogenesis of adhesive arachnoiditis is not clear. Burton19 suggested this to be the end point of an inflammatory process starting with radiculitis and progressing to arachnoiditis and adhesive arachnoiditis. As a result of inappropriate proliferation of arachnoid cells and production to dense collagen deposits surrounding nerve roots causes them to scar the meninges. Idris et al20 hypothesised an autoimmune-related mechanism which implicates nervous, immune and viscera musculoskeletal systems as important adjuncts in its pathophysiology, though its association with MCTD is yet to be evaluated. As mentioned before, a number of inflammatory insults have been associated with adhesive arachnoiditis. With our patient, underlying autoimmune connective tissue disease was possible inciting inflammatory event after ruling out all other possible causes on basis of laboratory data, neurophysiological studies and neuroimaging. True incidence of adhesive arachnoiditis and relationship to MCTD remains to be accurately documented.
Being a rare disorder, there is no consensus on standard treatment of adhesive arachnoiditis. To date, conservative approach (medications, physical therapy, psychotherapy, epidural steroid injections) and surgical treatment options are decided on a case-by-case basis with mixed clinical outcomes. In our case, likely autoimmune basis of our patient's presentation guided us towards augmentation of immunosuppressive therapy and addition of rituximab which worked out well in our patient.
Learning points
Mixed connective tissue disease (MCTD), a distant overlap syndrome, can eventually manifest symptoms more consistent with systemic lupus erythematosus (or scleroderma/polymyositis) and vice versa.
Adhesive arachnoiditis could be considered in an MCTD patient presenting with myeloradiculopathic symptoms.
Arachnoiditis can also mimic the symptoms of other diseases, such as cauda equine syndrome, spinal cord tumours, arachnoiditis ossificans and syringomyelia, so it is important to differentiate between arachnoiditis and other neurological manifestations of MCTD.
MRI and CT scans are important diagnostic investigations in suspected adhesive arachnoiditis.
Further studies will help to define the complex underlying pathophysiology of adhesive arachnoiditis, involvement of the nervous system and role of immune system as currently only limited publications are available to address this uncommon entity.
References
Footnotes
Contributors MUK was the chief author of the article and undertook most of the literature review. AF was the primary treating physician of the patient and was actively involved in subsequent revision of the article. JAJD consulted on the management of the patient.
Competing interests None declared.
Patient consent Obtained.
Provenance and peer review Not commissioned; externally peer reviewed.