Article Text

Summary
Juvenile hyaline fibromatosis (JHF) is a rare progressive autosomal recessive disease that is characterised by papulonodular skin lesions, soft tissue masses, joint contractures, gingival hypertrophy and osteolytic bone lesions. We present an 18-month-old boy with JHF. This case demonstrates that JHF should be considered in the differential diagnosis when multiple subcutaneous nodules are observed in the face, head and neck. Gum hypertrophy with palatal nodules is unusual in JHF.
Statistics from Altmetric.com
Background
Juvenile hyaline fibromatosis (JHF) is a rare hereditary disorder with mesenchymal dysplasia. The disease is characterised by multiple tumorous mucocutaneous proliferations, joint contractures, gingival hypertrophy and osteolytic lesions. It is hypothesised that JHF is a connective tissue disorder with a progressive nature. It requires repeated surgical interventions and physiotherapy. Rehabilitation and counselling of parents play an essential role in the treatment of these patients. Only about 70 cases of JHF have been reported worldwide to date. In the reported cases, nodules in the palate are very rare.
Case presentation
An 18-month-old boy born to second-degree consanguineous parents was referred to our institution for the treatment of multiple scalp swellings and gingival hyperplasia with the suspicion of leukaemia. He had motor developmental delay, flexion contractures of both hips, widening of the wrists (figure 1A), gingival hypertrophy, a nodular lesion over the palate and verrucae on the dorsum of the tongue and at the oral commissure (Figure 1B) and multiple subcutaneous nodules over the scalp (figure 1C) since 6 months of age. There was no history of a similar illness in the family members. The parents initially sought medical advice at 8 months of age in view of the painless progressive enlargement of the scalp nodules and gingiva. The patient was undernourished due to feeding difficulties related to the nodule in the palate.

(A) Widening of both wrists. (B) Gingival hypertrophy with a nodular lesion over the palate and verrucae on the dorsum of the tongue and at the oral commissure. (C) Multiple scalp nodules. (D) MRI head showing the subcutaneous nature of the nodules.
Investigations
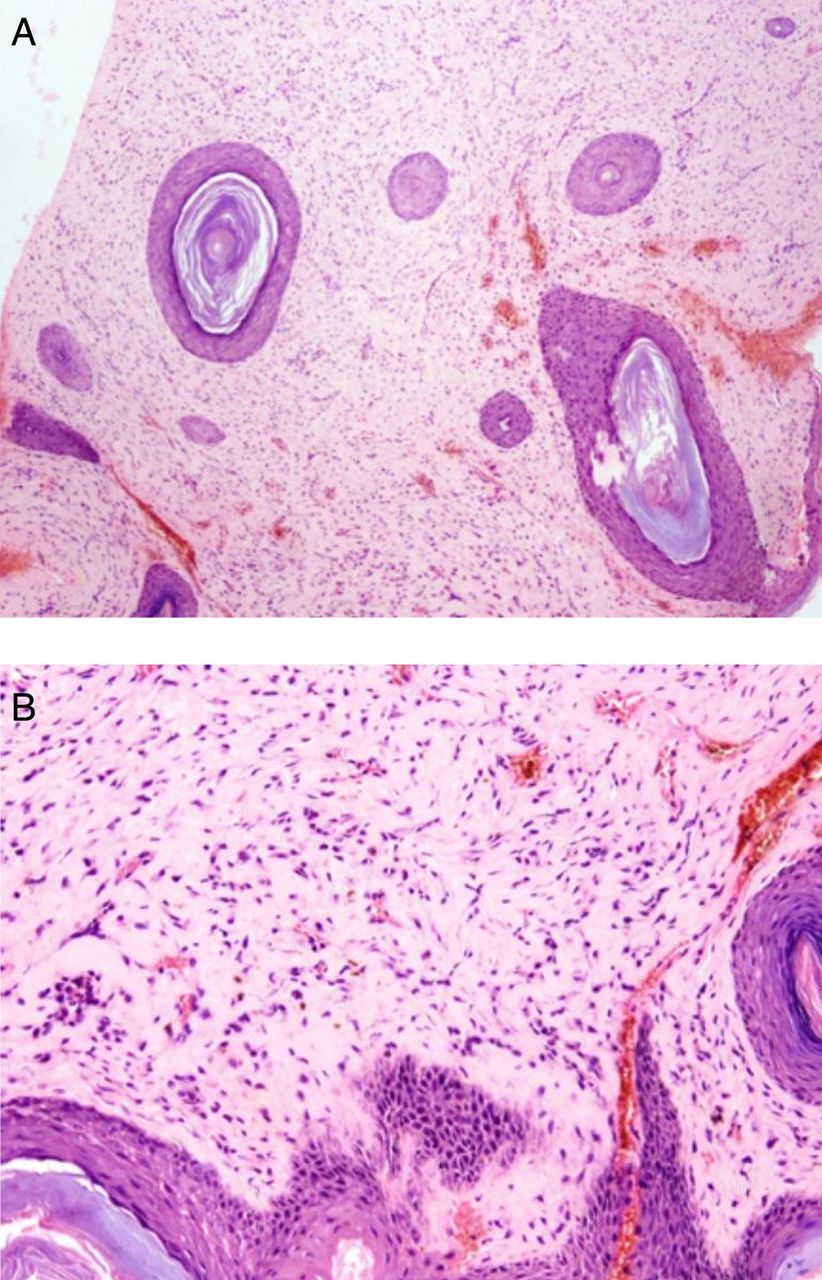
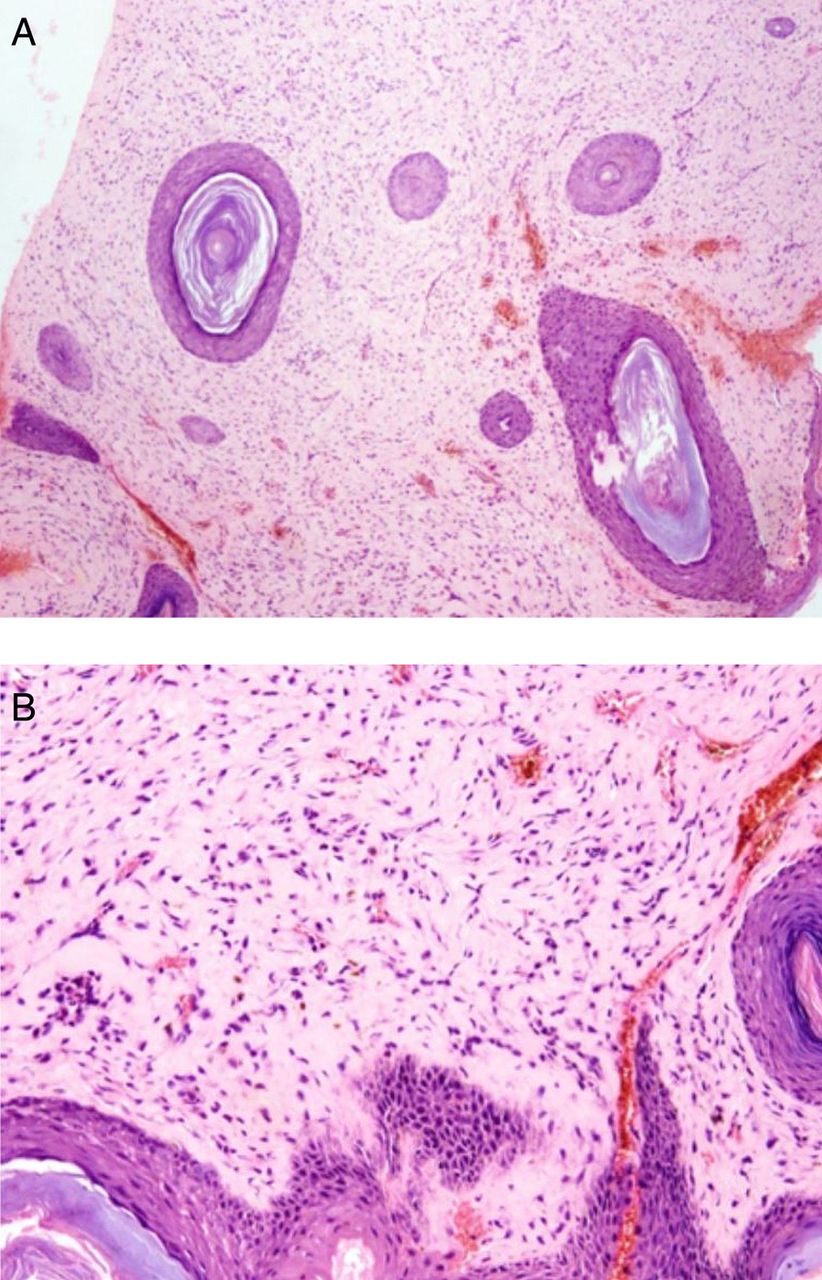
Laboratory examination and investigation of other organs were normal. A skeletal survey did not reveal any significant abnormalities. MRI of the head confirmed the subcutaneous nature of the scalp lesions (figure 1D). The subcutaneous nodule was surgically removed and fixed in 10% buffered formalin for 24 h. Whole tissue was embedded after routine processing. 3–5 µm sections were obtained and stained with H&E and with additional stains such as periodic acid Schiff (PAS), PAS after diastase, orcein (for elastic fibres) and Alcian blue. Biopsy of the nodule revealed markedly thickened dermis with abundant eosinophilic matrix (figure 2A) and uniform fibroblast-like cells embedded in an abundant eosinophilic ground substance (figure 2B). The material was PAS positive and diastase resistant. The material did not stain with Alcian blue and it lacked elastic fibres. A diagnosis of JHF was therefore made.
{kind=link}
{kind=link}
(A) Markedly thickened dermis with abundant eosinophilic matrix. (B) Uniform fibroblast-like cells embedded in an abundant eosinophilic ground substance.
Differential diagnosis
Gingival hypertrophy
Drugs (phenytoin, cyclosporine, nifedipine)
Lysosomal disorders (especially Farber's and I cell disease)
Infantile myofibromatosis
Mucopolysaccharidosis
Langerhan cell histiocytosis
Leukaemia
Tuberculosis
Sarcoidosis
Vitamin C deficiency
Plasma cell gingivitis
Subcutaneous nodules
Acute rheumatic fever
Juvenile idiopathic arthritis
Neuroblastoma
Acute myeloid leukaemia
Malignancies
Panniculitis
Granuloma annulare
Treatment
The parents were counselled regarding the nature of the disease. Surgical excision of the scalp lesions was advised but the parents did not bring the child for follow-up.
Outcome and follow-up
The patient was lost to follow-up.
Discussion
JHF is an autosomal recessive disease characterised by gum hypertrophy, joint contractures, bone lesions and internal organ involvement.1 It was first described as molluscum fibrosum by McMurray in 1873, later renamed as JHF by Kitano in 1972.2 It is characterised by deposition of amorphous hyaline material in the extracellular spaces of the dermis and soft tissues. Many case reports have reported different metabolic defects such as procollagen, tropocollagen, glycosaminoglycans, hyaluronic acid, chondroitin sulfate, type I, III and VI collagen.3 It arises from mutation in the anthrax toxin receptor 2 gene on chromosome 4q21.4
Although mucocutaneous nodules have been reported in the neck, elbow, knees, shins and ankles, a lesion in the palate has never been reported. Mucocutaneous lesions of the anal canal are associated with rectal bleeding.5 Affected children are mentally normal except for a few cases.6 Cosmetic and functional disability can be minimised by surgical excision of the lesions and physiotherapy to prevent joint contractures. The clinical criteria for resection of the lesions are functional impairment and ulceration of the lesion. Partial or radical gingivectomy followed by good oral hygiene can lead to resolution of hypertrophy; however, gingivectomy is usually followed by recurrence of the disorder.7 Although interferon α-2B has been tried in JHF,8 no definitive cure for JHF has yet been found. Other treatments for symptoms of joint involvement include cortisone, D-penicillamine and capsulotomy. Due to accumulation of abnormal materials in the soft tissues, severe airway problems can occur which may lead to anaesthetic complications.9 Death may occur due to pneumonia and respiratory failure due to peribronchial infiltrate and fibrous visceral pleural thickening.
Mutation, clinical features and histopathology are similar in infantile systemic hyalinosis (ISH) and JHF and therefore should be differentiated. JHF is a less severe late-onset disorder (after 3 months of age) and most children will survive until the fourth decade whereas ISH is a severe early-onset (few weeks of life) disorder which presents with short stature, failure to thrive, hyperpigmented plaques on bony prominences, persistent diarrhoea, visceral involvement, recurrent infections and death before 2 years of age.10
Since JHF and ISH share may common features, Nofal et al proposed the common term ‘hyaline fibromatosis syndrome’ with the following grading system:11
Grade 1: skin and/or gingival involvement only
Grade 2: grade 1 + joint and/or bone involvement
Grade 3: grade 2 + internal organ involvement with or without clinical manifestations
Grade 4: grade 3 + severe clinical decompensation (organ failure and/or septicaemia).
Our patient was grade 2 by clinical examination; however, complete assessment for involvement of internal organs was not done due to financial constraints and also because the parents were not willing.
Learning points
Early diagnosis of juvenile hyaline fibromatosis (JHF) can prevent cosmetic and functional disability.
Although manageable, recurrence after surgical excision is discouraging.
JHF should be considered in the differential diagnosis when multiple subcutaneous nodules are observed in the face, head and neck.
Gingival hypertrophy with palatal nodules is unusual in JHF.
Footnotes
Contributors PR prepared the manuscript. BK diagnosed the case, took clinical pictures and helped in preparing the manuscript. MV diagnosed the disease histopathologically. JXS diagnosed and managed the case, corrected the manuscript and helped in the review of the literature.
Competing interests None declared.
Patient consent Obtained.
Provenance and peer review Not commissioned; externally peer reviewed.