Article Text

Abstract
Hydroxychloroquine is a disease-modifying antirheumatic drug used for various rheumatological conditions. Its long-term use is well-known to have toxic effects on cardiac muscle cells. We present a biopsy-proven case of hydroxychloroquine-induced cardiotoxicity with detailed histopathological and imaging findings. The patient was referred to our heart failure clinic for concerns of reduction in left ventricular ejection fraction despite being on guideline-directed medical therapy. She had been diagnosed with rheumatoid arthritis, pulmonary hypertension and then subsequently heart failure with reduced ejection fraction 5 years ago. The evaluation included right heart catheterisation, cardiac MRI and endomyocardial biopsy. Light and electron microscopy showed myocyte hypertrophy and vacuolar change, abnormal mitochondria, myeloid bodies and curvilinear bodies. These findings were specific for hydroxychloroquine-induced cardiomyopathy. This case highlights the importance of clinical monitoring, early suspicion and consideration of drug-induced toxicities as a culprit for heart failure.
- Heart failure
- Cardiovascular system
- Rheumatoid arthritis
- Cardiovascular medicine
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
The sudden resurgence of hydroxychloroquine (HCQ) during the COVID-19 pandemic has brought the drug back to the limelight. As one of the most frequently prescribed disease-modifying antirheumatic drugs (DMARDs), HCQ remains an integral part of treatment for a myriad of rheumatological conditions. The known cardiac manifestations of HCQ-induced toxicity include conduction abnormalities, ventricular hypertrophy, hypokinesia and lastly, cardiomyopathy. HCQ-induced cardiomyopathy is poorly understood and remains underdiagnosed. Due to its lipophilic properties, HCQ can permeate lysosomes of myocytes, and by binding phospholipids and glycogen, causes an acquired storage disorder that leads to fibrillar disorganisation, atrophy and fibrosis.1 Diagnosis of this potentially reversible condition requires a high degree of suspicion with the right set of tools to rule out other more common aetiologies. Timely withdrawal of the offending drug can halt and potentially reverse cardiomyopathy.2
Case presentation
A woman in her 60s was referred to the heart failure clinic for concerns of worsening dyspnoea and fatigue, which impaired her activities of daily living. Her symptoms had been ongoing for the last 5 years but had gotten worse over the last few months. Her medical history was significant for New York Heart Association class III, American College of Cardiology stage C heart failure with reduced ejection fraction (HFrEF) secondary to non-ischaemic cardiomyopathy, non-insulin-dependent type II diabetes mellitus, hyperlipidaemia, hypertension, obstructive sleep apnoea, rheumatoid arthritis (RA) with associated pulmonary nodulosis and Sjogren’s syndrome. A prior echocardiogram from 5 years ago showed left ventricular ejection fraction (LVEF) of 40%–45%, mild global hypokinesis, mild concentric left ventricular hypertrophy and no significant valvular findings. Left heart catheterisation that was performed at the time of initial diagnosis 5 years ago had shown no significant coronary artery disease. The patient had been started on guideline-directed medical therapy (GDMT) 5 years ago and was on carvedilol 12.5 mg two times per day, sacubitril/valsartan 24–26 mg two times per day, spironolactone 25 mg once daily and dapagliflozin 10 mg once daily. Dose escalation had been limited due to symptomatic hypotension in the past. Her other medications included bumetanide 1 mg daily, aspirin 81 mg daily, leflunomide 20 mg daily, HCQ 400 mg daily, prednisone 5 mg daily, and rituximab 1000 mg intravenously, every 4 months.
On presentation, vital signs included a blood pressure of 105/56 mm Hg while sitting, and a heart rate of 61 beats/min. Pertinent physical examination findings included a jugular venous pressure of 10 cm, a positive hepatojugular reflex and bilateral pitting oedema up to her knees. No wheezing was heard on auscultation of her lungs, but crackles were heard at bilateral lung bases and cardiac examination revealed normal S1 and S2 with no murmurs.
Investigations
The evaluation began outpatient with laboratory work that was unremarkable except for a B-type natriuretic peptide of 441 pg/mL. ECG showed a normal sinus rhythm with a new left bundle branch block. Repeat echocardiography was performed and showed a depressed LVEF of 25%–30%, mild to moderate mitral regurgitation, moderate right ventricular dilatation, septal flattening throughout the cycle, biatrial dilatation and a raised pulmonary artery systolic pressure of 75 mm Hg. Due to concern for pulmonary hypertension (PAH), right heart catheterisation was scheduled for further haemodynamic evaluation, which revealed right atrial pressure 14 mm Hg, right ventricular pressure 90/20 mm Hg, significantly elevated pulmonary artery pressure 90/38 mm Hg with a mean of 55 mm Hg, elevated pulmonary capillary wedge pressure 20 mm Hg and a preserved cardiac index. These findings showed elevated right and left-sided filling pressures with pulmonary artery hypertension. The patient was then admitted for further evaluation.
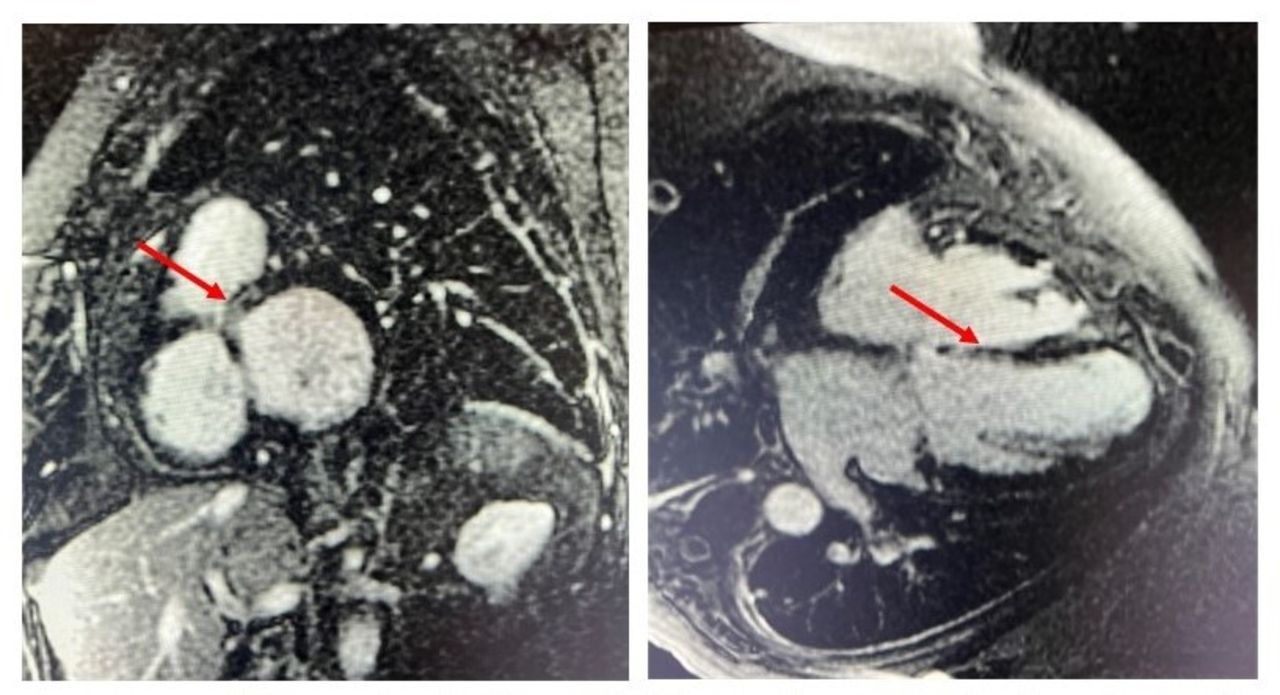
Based on the patient’s history, there was low suspicion for ischaemia leading to the current clinical presentation. A cardiac MRI (CMRI) was obtained on admission that showed no subendocardial delayed gadolinium enhancement (DGE) that further supported that this new reduction in ejection fraction was not driven by ischaemia. However, CMRI did show mesocardiac DGE involving the interventricular septum, particularly prominent in the basal septal and inferoseptal segments (figure 1). There were no other areas of DGE.

Cardiac MRI showing late gadolinium enhancement in the mesocardium of the basal segment of interventricular septum (red arrows).
Her initial testing also included repeat imaging of her lung parenchyma with a high-resolution CT scan given her history of pulmonary nodules. It revealed numerous cavitary nodules compatible with rheumatoid nodules, bilateral ground-glass opacities with central bronchial wall thickening and interlobular septal thickening compatible with pulmonary oedema. The patient was also found to have trace pleural effusions with associated pleural thickening, mediastinal and bilateral hilar lymph nodes. A ventilation-perfusion (VQ) scan was ordered to rule out prior pulmonary embolism that might be contributing to the PAH. It showed multiple segmental and subsegmental perfusion defects predominantly involving the bilateral upper lobes but no ventilatory defects.
Pulmonology and rheumatology expertise were sought to assist with the workup. Her severe PAH was thought to be driven by a combination of WHO group I due to her known RA, group II based on the right heart catheterisation and group IV based on the high probability VQ scan findings. After discussion, the patient was started on lifelong anticoagulation with warfarin. Hypercoagulability workup later revealed that the patient had a heterozygous mutation of factor V Leiden that is associated with a 5-fold to 10-fold increase of venous thrombosis.
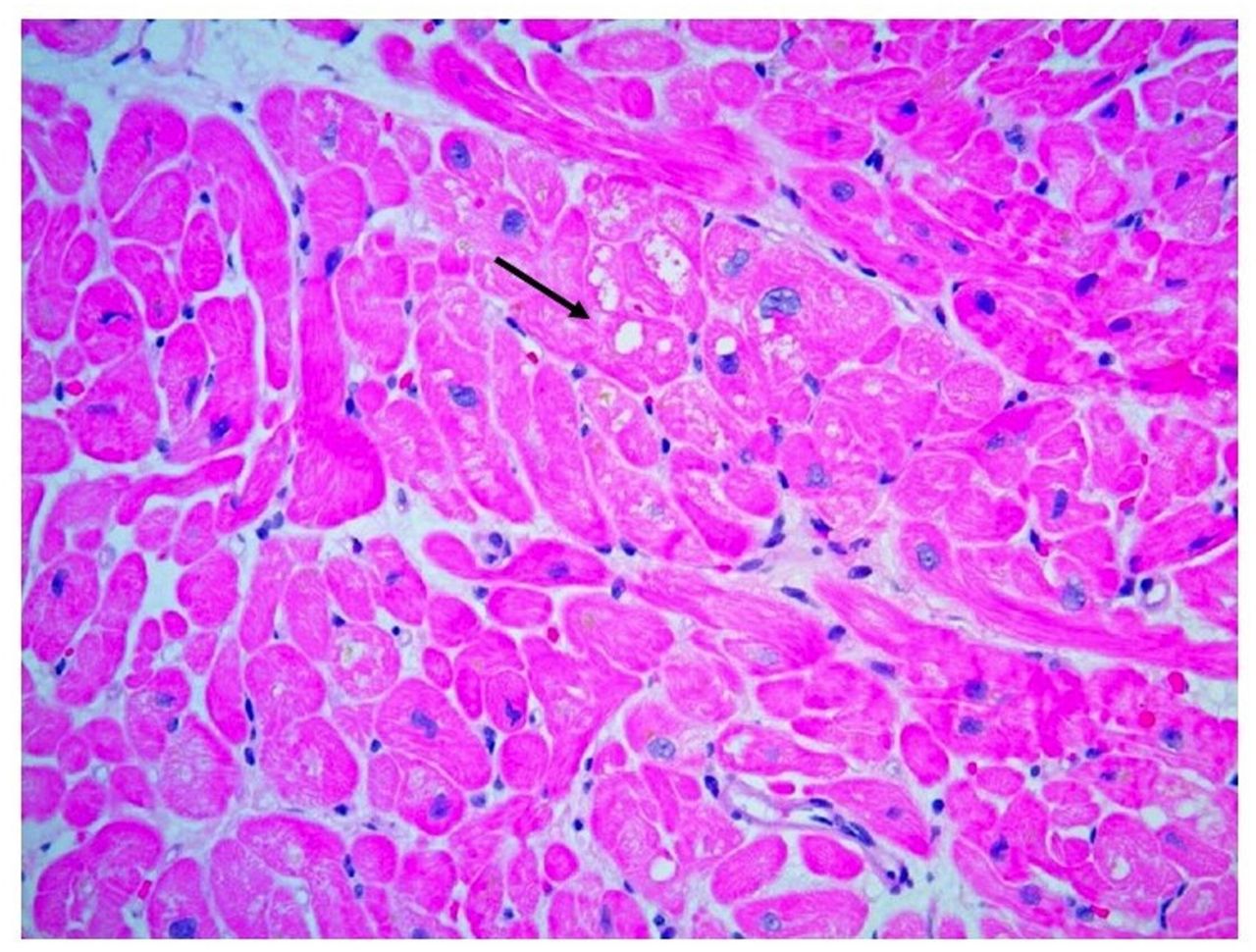
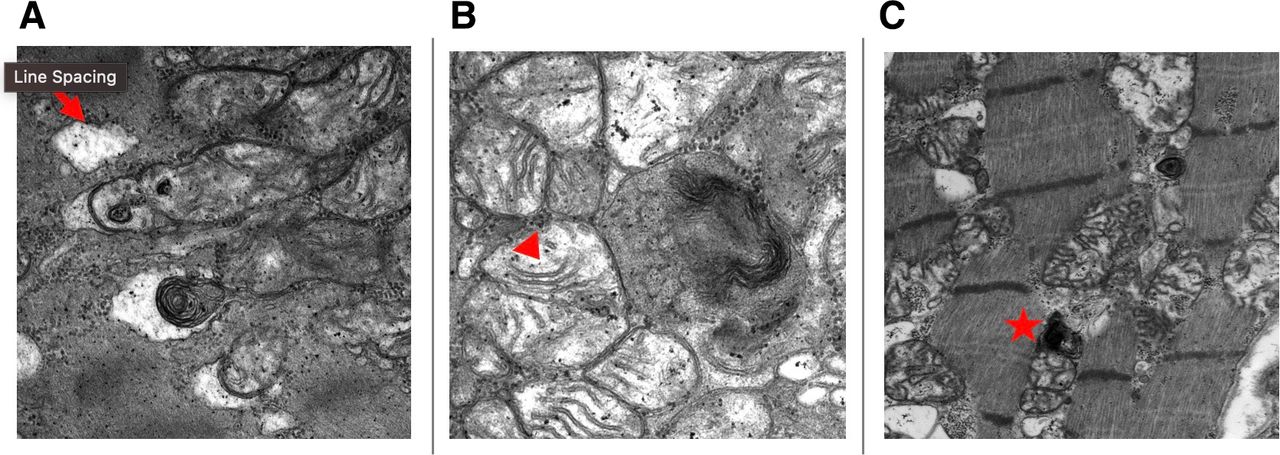
Despite above investigations, the aetiology of her HFrEF remained undetermined. At this point, the patient had been on HCQ for 6 years and suspicion rose that it might be contributing to the cardiomyopathy. Her CMRI findings were also pointing toward an infiltrative process and a decision was made to pursue an endomyocardial biopsy (EMB). The H&E-stained slides showed myocyte hypertrophy and mild, patchy myocyte vacuolar change with no significant inflammation or myocardial fibrosis (figure 2). Electron microscopy showed numerous abnormal mitochondria with enlargement and disordered cristae, myeloid bodies and curvilinear bodies (figure 3). Biopsy findings were consistent with HCQ toxicity.

Light microscopy showing myocyte hypertrophy and vacuolar changes without significant interstitial fibrosis.

{kind=link}
{kind=link}
{kind=link}
Electron microscopy showing (A) vacuolar degeneration (arrow), (B) abnormal mitochondria with enlargement and disordered cristae (arrowhead), (C) abnormal mitochondria with curvilinear bodies (star).
Differential diagnosis
The differentials for diagnosis of HCQ toxicity in the heart are broad and include storage disorders like Fabry disease and adult-onset Pompe disease (acid maltase deficiency), drug-induced myopathy (amiodarone, rituximab, prednisone, cocaine, cobalt and perhexiline maleate), chemotherapeutic agents, genetic diseases (Danon disease and other mitochondrial disorders) and ischaemic heart disease.3
Treatment
HCQ was discontinued and the patient was discharged. The patient was continued on her GDMT, with plans of outpatient cardiac resynchronisation therapy with defibrillator placement, follow-up with cardiology to monitor symptoms and follow-up with pulmonology to initiate pulmonary vasodilators.
Outcome and follow-up
The patient was initiated on riociguat 2.5 mg three times a day and treprostinil 12 breaths four times daily for her PAH management. A repeat echocardiogram was performed outpatient 6 months after discontinuation of HCQ, with no significant change in contractility on a side-to-side comparison of the echocardiogram performed before her hospitalisation. However, there was an interval decrease in her estimated pulmonary artery systolic pressure, which came down to 64 mm Hg. Per the patient’s report, her symptoms improved and her exercise capacity increased.
Discussion
HCQ and chloroquine have a recognised role as DMARDs in the treatment of RA, Sjogren’s disease and systemic lupus erythematosus. The chronic nature of these diseases leads to long-term exposure to DMARDs, and the resultant drug accumulation predisposes patients to their adverse effects. The rheumatological properties of HCQ have been attributed to the neutralisation of antigen processing in antigen-presenting cells but its exact mechanism is unknown.4 HCQ is absorbed rapidly and gets accumulated in various tissues. High concentrations are found in the liver, lungs, kidneys, eyes, skeletal muscles and heart tissue.5 As a result of longitudinal exposure and accumulation, multiple cardiac manifestations of these drugs have been identified, the most prominent being electrophysiological disturbances. Drug-induced cardiomyopathy remains an important and under-recognised aetiology of cardiomyopathy and heart failure.
Risk factors for the development of HCQ-induced cardiotoxicity are thought to include older age, female sex, longer duration of therapy (>10 years), elevated per-kilogram daily dose, pre-existing cardiac disease and renal insufficiency.6 Symptom onset and disease progression can be variable, ranging from 3 months to 27 years with a mean of 10 years.7 A wide range of cumulative dosage of antimalarial drugs in heart failure cases has been noted (270–9125 g). A safe daily dose where only reversible asymptomatic pigment changes in the retina were observed was defined as 6.0–6.5 mg/kg/day for HCQ.8 Our patient who weighed about 110 kg had been on HCQ for 6 years prior to diagnosis of HCQ-induced cardiotoxicity, and her total cumulative dosing was 876 g by then. It is important to note that the patient started to develop symptoms of heart failure only a year after she had been on HCQ.
HCQ crosses the cell membrane and preferentially binds to the phospholipids, thus causing inhibition of phospholipases inside cardiac myocytes. This leads to an acquired lysosomal storage disorder due to the inhibition of constitutive autophagy present in normal cardiac myocytes. The accumulation of glycogen and phospholipids leads to an acquired, drug-induced glycogen storage disorder, which presents itself as lamellar inclusion bodies and curvilinear bodies in cytoplasm that can be visualised with electron microscopy. Ultimately, these changes result in concentric hypertrophy along with the development of restrictive cardiomyopathy, which eventually leads to heart failure.
Due to similar mechanisms, drug-induced phospholipidosis can mimic its close differential Fabry disease. Myeloid bodies (also referred to as zebra bodies) can be seen in HCQ-mediated cardiotoxicity, amiodarone toxicity as well as Anderson-Fabry disease. As seen in our patient, the presence of curvilinear bodies is thought to be a specific finding that differentiates HCQ-induced lysosomal storage disorder from Anderson-Fabry disease.9 10 Our review did show a case of HCQ-mediated lysosomal store disorder without characteristic curvilinear bodies on renal biopsy.10 Therefore, a carefully obtained personal, as well as family history and age of onset, can play a key role in differentiating the two disorders. In this case, apart from HCQ, the patient was also on leflunomide, prednisone and rituximab, all of which can rarely cause non-ischaemic cardiomyopathy and were considered during evaluation. The electron microscopy findings, therefore, played a crucial role in identifying the definitive diagnosis. These histopathological findings may unfortunately persist for years after drug discontinuation.11 This may be the reason why the ejection fraction has not improved in our patient at the time of 6-month follow-up. While there is a paucity of literature investigating PAH in RA, the pooled prevalence of PAH in RA has been reported as 22.3% by Yang et al based on review of two studies.12 13 It is also important to note that RA is associated with a higher prevalence of symptomatic heart failure. A retrospective incidence cohort study estimated excessive congestive heart failure risk in patients with RA (HR 1.87, 95% CI 1.47–2.39) after adjusting for demographics, ischaemic heart disease and cardiovascular risk factors.14 Our patient had a reduction of LVEF that was found to be secondary to HCQ-mediated cardiotoxicity. This suggests that a patient may have multiple underlying pathologies contributing to their illness and that HCQ-mediated cardiomyopathy should be kept in mind while evaluating patients who have underlying RA and are on HCQ.
EMB can help to rule out other infiltrative diseases such as amyloidosis and sarcoidosis as well as to differentiate between inherited lysosomal disorders and HCQ-induced lysosomal storage disorder. The potentially reversible nature of cardiomyopathy makes early diagnosis and discontinuation of the offending drug as the mainstay of treatment. Prognosis can be extremely variable, ranging from death, cardiac transplant or even partial or complete recovery.15 In the review by Costedoat-Chalumeau et al, which described 25 cases of heart failure, death occurred in 11 (46%) and partial or complete improvement was seen in 8 out of 12 cases where the drug was discontinued.7 In closing, high degree of suspicion, and timely utilisation of CMRI coupled with an EMB are the crucial diagnostic tools for clinching the evasive diagnosis of HCQ-induced cardiomyopathy.4
Learning points
Cardiomyopathy is a potentially reversible adverse effect of hydroxychloroquine (HCQ) therapy. Due to its similarity with other lysosomal storage disorders as well as limited number of cases, the diagnosis remains under-reported and underdiagnosed.
A high degree of suspicion, followed by cardiac MRI (CMRI) and ultimately endomyocardial biopsy (EMB) with electron microscopy to look for specific findings of HCQ-induced cardiotoxicity, is required to make this rare and evasive diagnosis.
In addition to ophthalmological screening, annual ECG as well as echocardiography screening for patients on long-term HCQ therapy should be considered with any abnormalities being followed up with CMRI and EMB.
Ethics statements
Patient consent for publication
References
Footnotes
Twitter @AnureetMalhotra, @DaliaTarun
Contributors AM and MAP collected the relevant data, reviewed current literature and drafted the manuscript, including designing the figures, while TD and AV revised the article and provided final approval for publication. All authors contributed to the study design, critically reviewed the first draft, approved the final version and agreed to be accountable for the work.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Case reports provide a valuable learning resource for the scientific community and can indicate areas of interest for future research. They should not be used in isolation to guide treatment choices or public health policy.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.