Article Text

Abstract
Haemophagocytic lymphohistiocytosis (HLH) is an aggressive hyperinflammatory haematological condition often associated with malignancy, infection or rheumatological disorders. HLH has rarely been associated with medications, including antibiotics. We describe a case of a patient without significant medical history who presented with HLH following treatment with trimethoprim/sulfamethoxazole (TMP/SMX). Additionally, we will discuss the possible mechanism of medication-induced HLH as well as the successful use of dexamethasone as the sole treatment. Early diagnosis and treatment of this disease is critical and medication-induced HLH should be considered in cases without a clear aetiology. To our knowledge, this is the first case report of TMP/SMX-induced HLH that was successfully treated with steroid monotherapy and just the second case report of TMP/SMX-induced HLH.
- Drugs: infectious diseases
- Haematology (incl blood transfusion)
- Pharmacology and therapeutics
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
Haemophagocytic lymphohistiocytosis (HLH) is a hyperinflammatory haematological condition that is often aggressive, particularly when not identified early. The mortality rate in adult patients is 26.5%–74.8%.1 The broad constellation of symptoms associated with HLH is similar to numerous other haematological, infectious, autoimmune and allergic aetiologies and can be easily misattributed to another condition. While HLH is rare, early identification and prompt initiation of treatment are critical. Recent guidelines describe patients with HLH syndrome as to encompass those who meet the HLH diagnostic criteria while defining patients with HLH disease as those in whom the immune dysregulation is their primary disease process.2 Optimal treatment, including the necessity for immune suppression, may differ depending on the aetiology and classification of HLH. The seminal HLH-94 and HLH-2004 trials provide the framework for how HLH is diagnosed and treated today.3 4 Our report presents the case of a patient who presented with multisystem organ failure shortly after beginning a course of trimethoprim/sulfamethoxazole (TMP/SMX) and was subsequently diagnosed with HLH. Medication-induced HLH is a rare entity without established protocols for treatment or large-scale reports of patient outcomes.
Case presentation
A male in his early 30s underwent an office-based incision and drainage of a perirectal abscess and was started on a 7-day course of TMP/SMX. On the second day following the procedure, he developed fever, chills, night sweats and a rash, which persisted and worsened until his presentation to the hospital 5 days later. On arrival to the emergency department, the patient was found to have a fever of 40.2°C (104.4°F). On physical examination, he was well appearing with a a faint, blanching and morbilliform rash on his trunk with extension to the proximal extremities. The perirectal abscess had largely resolved with no visible erythema or drainage and minimal scabbing and tenderness.
Investigations
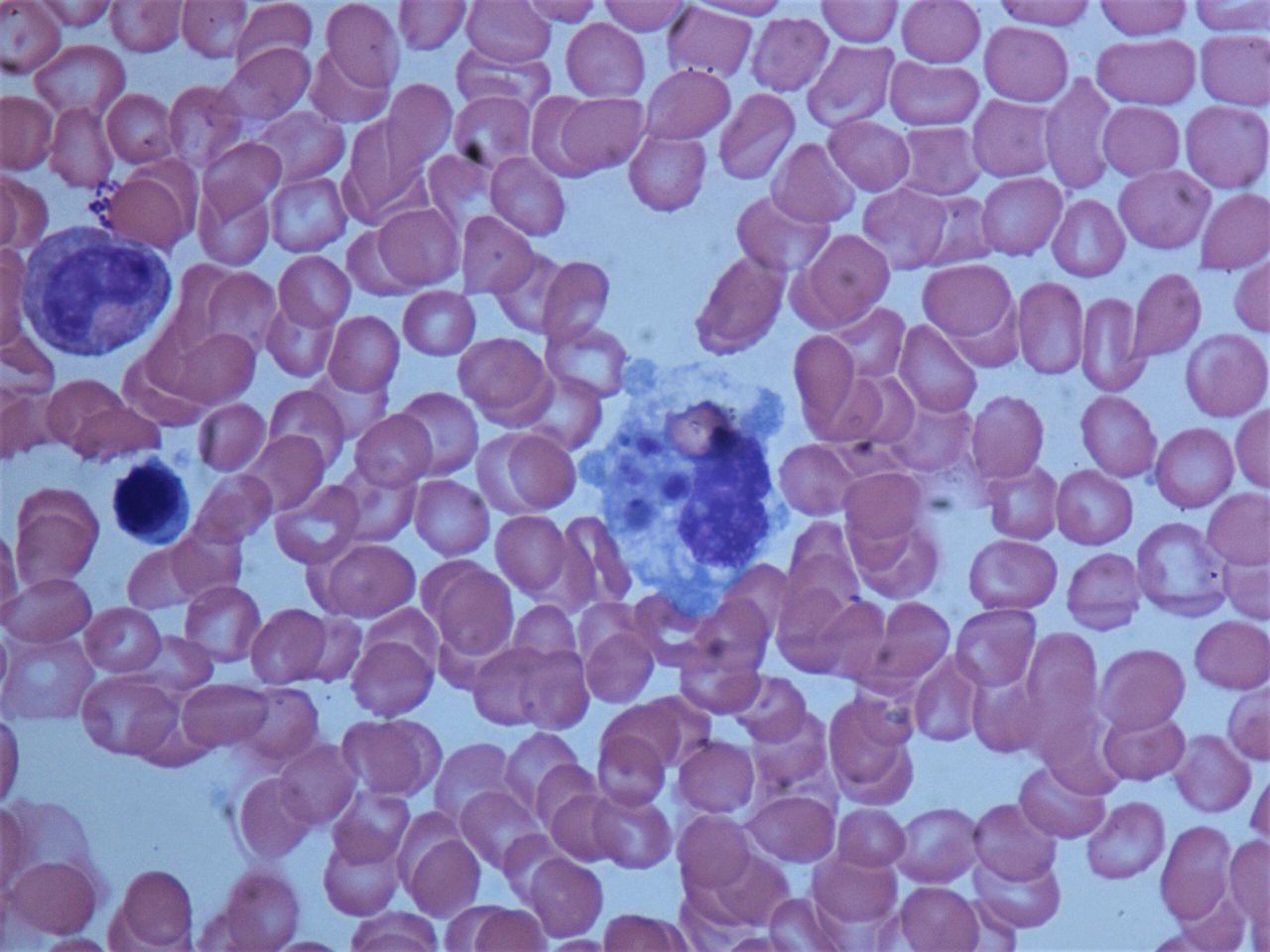
Initial laboratory testing showed pancytopenia (white cell count 1.29 x 109/L, haemoglobin 12.0 g/dL, platelets 89 x 109/L). He had significantly elevated inflammatory markers (D-dimer 25 375 ng/mL and ferritin 19 350 ng/mL). He also had mild acute kidney injury, elevated liver enzymes and coagulopathy. Over subsequent days, many of these laboratory abnormalities significantly worsened, including the pancytopenia and aspartate aminotransferase and alanine aminotransferase levels. He had rising inflammatory markers and development of hypertriglyceridaemia (table 1). The initial peripheral smear showed rouleaux formation, rare schistocytes and atypical lymphocytes, with no blasts or dysplastic leucocytes. A CT chest/abdomen/pelvis with intravenous contrast showed hepatosplenomegaly without any significant lymphadenopathy. A bone marrow aspirate with immunohistochemical stains for CD163 showed increased macrophages with haemophagocytosis (figure 1).
Selected laboratory values
{kind=link}
Haemophagocytosis in the bone marrow biopsy from our patient.
Differential diagnosis
The differential diagnosis included adult-onset Still’s disease, drug hypersensitivity syndrome or HLH syndrome secondary to an infectious, rheumatological or malignant condition.
Given the patient’s high fevers, elevated inflammatory markers and recent history of infection, infectious aetiologies for his presentation were initially considered as either the primary aetiology of the patient’s symptoms or as the trigger for HLH. The patient’s perirectal abscess had largely resolved and high-resolution contrast-enhanced CT imaging showed resolving inflammation. Blood cultures were drawn and serologies were sent for numerous viral, tick-borne and sexually transmitted illnesses including the following: 20 virus respiratory panel with COVID-19, IgM and IgG for Rocky Mountain Spotted Fever, Ehrlichia, Anaplasma and Babesia, Epstein-Barr virus (EBV) IgM antibody, EBV DNA quantitative PCR, Cytomegalovirus DNA quantitative PCR, HIV fourth generation assay and viral load, Neisseria gonorrhoeae and Chlamydia trachomatis nucleic acid amplification, hepatitis B surface antigen, hepatitis B core IgM, hepatitis A IgM. No infectious aetiology was identified by molecular diagnostic tests or cultures.
Once an infectious aetiology was ruled out, adult-onset Still’s disease was considered in light of his systemic illness, rash, elevated inflammatory markers and hepatosplenomegaly. Although our patient had pancytopenia, he did not have arthralgias or myalgias, which are often considered universal features of this disease.5 A type III or type IV hypersensitivity reaction including serum sickness, drug reaction with eosinophilia and systemic symptoms (DRESS), or Stevens-Johnson syndrome were possibilities as well, although the patient’s relatively benign rash would have been atypical. A haematological/oncological condition, including leukaemia or other malignancy, precipitating HLH was an initial possibility given the patient’s pancytopenia and hepatosplenomegaly; however, the bone marrow biopsy was negative for malignancy.
Our patient met 6 of 8 HLH-2004 criteria with an H-score of 239 consistent with a 98%–99% probability of HLH. Hereditary or genetic causes of HLH are another potential cause, commonly referred to as primary HLH. Testing for natural killer cell function, perforin expression and CD107a expression tests can be done in conjunction with mutation analysis to help identify these patients. However, given this patient’s age, lack of family history, relatively mild disease and rapid response to treatment, primary aetiologies were considered less likely.6 The negative workup for other secondary causes as well as a concomitant temporal association with the use of a new medication, and a presentation largely consistent with a systemic drug hypersensitivity reaction, made the diagnosis of HLH in this patient the most conceivable, likely triggered by the TMP/SMX itself.
Treatment
The patient was started on 10 mg/m2 intravenous dexamethasone for presumed HLH. Soon after initiating steroids, he defervesced and clinically improved. Laboratory markers of inflammation trended down and his acute kidney injury and cytopenia resolved. He was discharged on a slow steroid taper with close outpatient follow-up for continued monitoring.
Outcome and follow-up
Five months following initial presentation, the laboratory markers of HLH had all resolved, with the exception of a persistent mild indirect hyperbilirubinaemia. Steroids were discontinued. No malignancy or other systemic condition were identified during the workup. The patient remains asymptomatic and has returned to his prior baseline.
Discussion
In this case, we described a patient who developed HLH after taking TMP/SMX. Patients with HLH are typically treated as per the HLH-94 protocol, with dexamethasone, etoposide, methotrexate and cyclosporine used as mainstays of treatment.3 More recently, alemtuzumab has been used in cases of refractory HLH.7 Given the patient’s nontoxic appearance as well as initial positive response to steroids, he was not given etoposide or any other medication and responded well to a prolonged steroid taper.
The causative agent for HLH in this patient was likely TMP/SMX given the time course from the offending agent, no use of other medications, no other significant medical history and no underlying malignancy or other systemic condition identified. Medication-induced HLH is a rare entity with few prior published case reports and even fewer associated with antibiotics.8–13 The mechanism of the association between medications and HLH is unclear and there are no established guidelines for treatment of these patients. Patients with medication-induced HLH were not specifically included in the HLH-94 or HLH-2004 trials. The mechanism may be related to a drug hypersensitivity reaction leading to a CD8+T cell activation with a hyperinflammatory macrophage response which may lead to haemophagocytosis.8–10 Of note, sulfonamides have been identified as being particularly immunogenic in general.14 Additionally, TMP/SMX has been associated with the development of various haematological dyscrasias, including immune thrombocytopenic purpura and agranulocytosis.15 16 There is support that medication-induced HLH is related to a drug hypersensitivity reaction and prior reports have noted similarities with DRESS. However, eosinophilia is not seen in HLH and excessive CD8+T cell activation or haemophagocytosis would not be expected in DRESS.12 Prior case reports have noted quick response to therapy and relatively favourable outcomes in medication-induced HLH in comparison to more common aetiologies of HLH syndrome.10 Our patient’s rapid and lasting response to steroids alone may also be indicative of medication-induced HLH.
Patient’s perspective
As a relatively healthy young man, I was obviously caught by surprise by HLH. Looking at my blood results and seeing so many abnormal levels I couldn’t reconcile what I felt with what was being reported through my lab results. Aside for a fever and some fatigue, there wasn’t really any pain, but I was told I was suffering from multi-system organ failure. Now that it’s all behind me it’s still hard to believe that it happened. And of course, I am grateful for the great care and treatment given to me by my medical providers.
Learning points
Haemophagocytic lymphohistiocytosis (HLH) is a rare hyperinflammatory haematologic condition often associated with malignancy, infection or rheumatological disorders.
It is crucial to identify and diagnose HLH early so that proper treatment can be initiated, both for HLH and the underlying condition, if present.
HLH has rarely been associated with medications, including antibiotics, but should be considered as a possibility in patients with HLH without an identifiable primary condition.
Medication-induced HLH is likely to be related to a drug hypersensitivity reaction which may respond well to treatment with steroid monotherapy.
Ethics statements
Patient consent for publication
References
Footnotes
Contributors AZ and IH contributed to drafting and editing this manuscript.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Case reports provide a valuable learning resource for the scientific community and can indicate areas of interest for future research. They should not be used in isolation to guide treatment choices or public health policy.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.