Article Text

Abstract
Axial myopathies with paraspinal predominance usually present with dropped head, abnormal posture or rigidity of the spine. Management of axial myopathy can be difficult and there is little data in the literature about surgical treatment. We discuss a case of axial myopathy with late-onset scoliosis and dropped head, focusing on the surgical management of the case.
- muscle disease
- neuroimaging
- neuromuscular disease
- spinal cord
- neurosurgery
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
Adult onset axial myopathies can be classified into muscle disorders with predominant axial involvement with or without more widespread muscle involvement. Clinical manifestations can include head drop, bent spine syndrome, late-onset scoliosis and rigid spine syndrome. Potential causes include isolated neck extensor myopathy, inflammatory myopathies including the brachiocervical inflammatory myopathy, sporadic late-onset nemaline myopathy and a range of genetic myopathies.1 Recognition of axial myopathies can be challenging, but spinal MRI imaging can complement clinical judgement by identifying (sometimes subtle) oedema and fatty infiltration of paraspinal muscles. Paraspinal muscles are rarely sampled and abnormalities may be seen even in healthy subjects, so any abnormalities need to be interpreted with caution. Management of axial myopathies can be difficult. We describe a case of a late-onset axial myopathy, where significant surgical intervention resulted in a good outcome for the patient.
Case presentation
A 76-year-old Polish nun was referred to our neuromuscular unit with a 1 year history of head drop. Three years prior to this, she was seen by the spinal surgeons following a 1 year history of progressive thoracolumbar scoliosis. Examination at the time revealed marked truncal shift to the right and neurological assessment was unremarkable. Due to significant compromise to quality of life, a two-stage scoliosis correction procedure with instrumented fusion from T2 to L4 was performed. The first surgical stage involved an anterior release with a standard thoracotomy and retroperitoneal approach to the spine. The second stage with posterior instrumented fusion although initially planned for a week later, had to be postponed by a month to allow appropriate recovery. Four weeks post-operatively her saggital and coronal balance had improved and height had increased by 1.5 cm. One year later, she had resumed her daily activities and felt like she had regained her health.
Two years post-operatively she developed a head drop with associated discomfort of shoulder and neck muscles, and was subsequently referred to our service. She did not have any bulbar or other neuromuscular symptoms. There was no relevant family history of neuromuscular disease. Examination revealed weak neck extensor muscles with paraspinal muscle atrophy and wasting around shoulder girdle. There were no fasciculations or fatigable muscle weakness. Cranial nerve examination was normal and there were no long tract or extrapyramidal signs. The iliopsoas and quadriceps muscles were mildly weak (MRC grade 4+) bilaterally.
After unsuccessful conservative management of head drop, extension into the cervical spine was considered. Our patient was very motivated for surgery, was in reasonable health and fortunately there were no major surgical complications.
Investigations
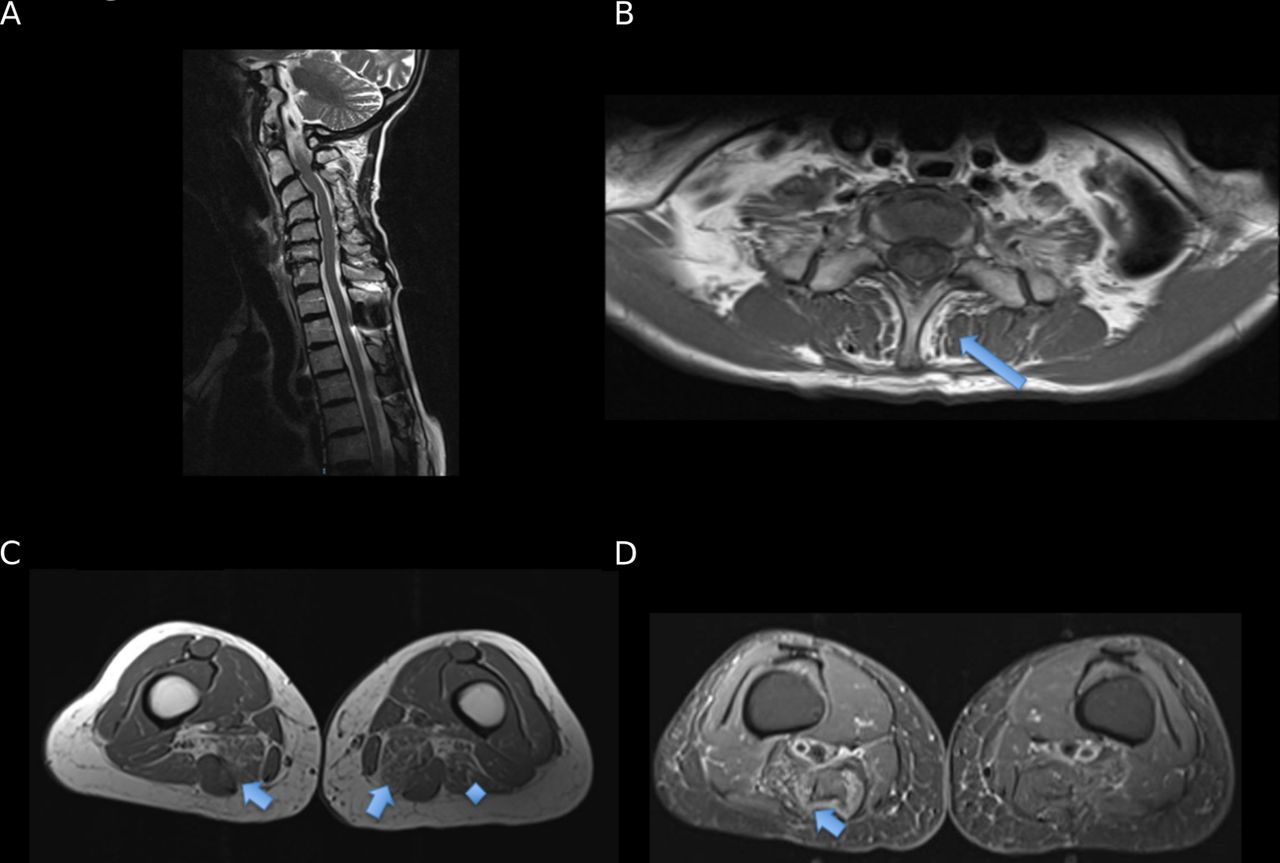
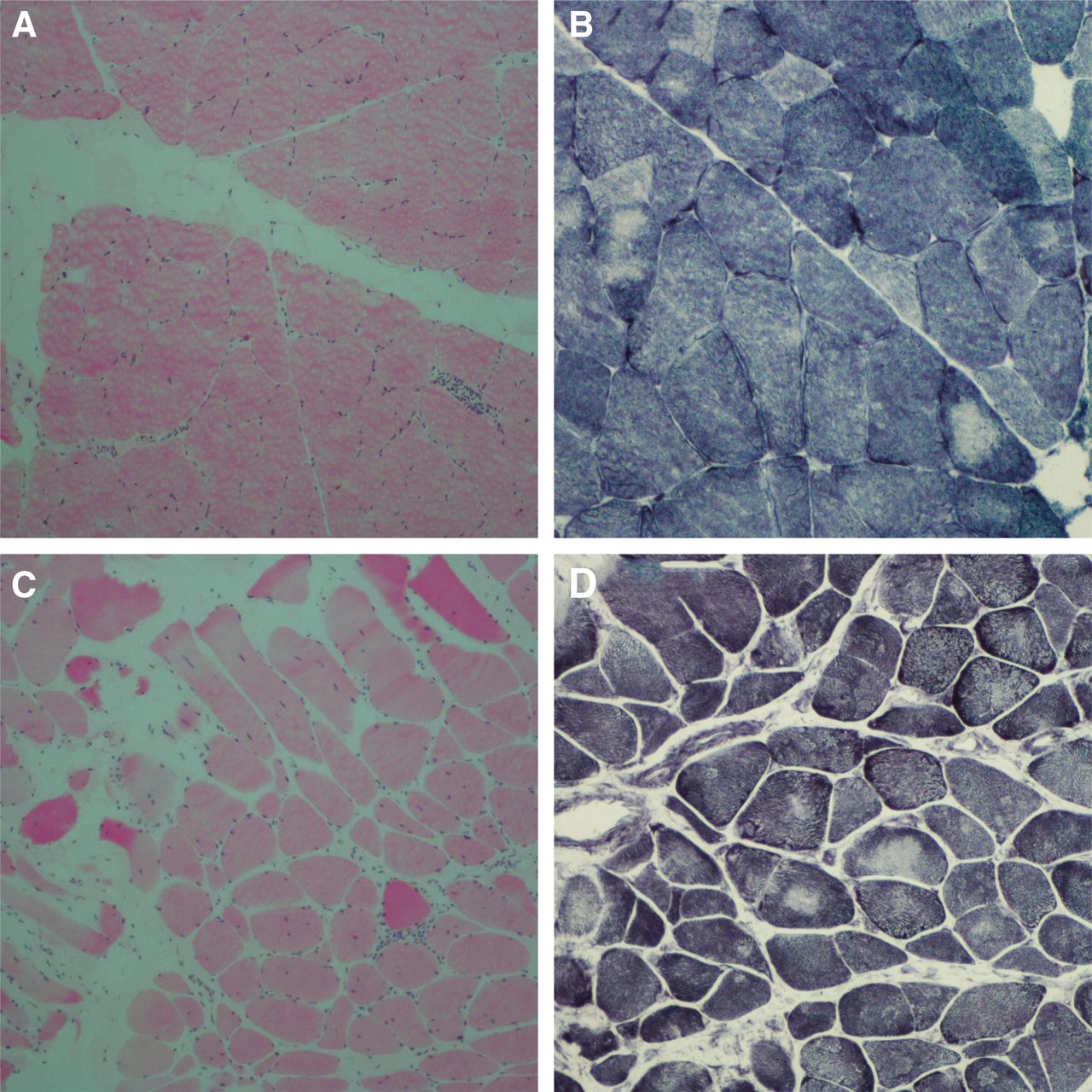
Initial investigations revealed a creatine kinase level of 167 IU/L (normal range 25–200), thyroid function and parathyroid hormone. An autoimmune profile including myositis, acetylcholine receptor (AChR) and muscle-specific kinase (MuSK) antibodies were all negative. MRI of cervical spine (figure 1A) revealed multilevel degenerative disc disease with no associated cord signal change lung function tests and 12 lead ECG were normal. There was oedema and fatty atrophy within posterior cervical paraspinal musculature (figure 1B). MRI scan of thighs showed oedema and fatty infiltration of left biceps femoris muscle and semimembranosus and tibialis anterior muscles bilaterally (figure 1C,D). Nerve conduction and repetitive stimulation tests were normal. Needle electromyography (EMG) showed spontaneous activity in tibialis anterior muscles bilaterally and small polyphasic motor units in a patchy pattern, particularly involving the cervical paraspinal muscles. Cervical paraspinal and right quadriceps muscle biopsies (figure 2) showed myopathic features with mild inflammatory changes predominance of slow fibres, several cores and a few cytochrome deficient/succinate dehydrogenase positive muscle fibres consistent with her age without any ragged red fibres. Targeted genetic testing for ryanodine receptor 1 (RYR1), facioscapulohumeral muscular dystrophy and myotonic dystrophy type 2 were all negative. Subsequent whole exome sequencing did not identify any primary finding to account for her clinical phenotype.

(A) Sagittal T2w midline image of cervical spine showing anteriolithesis of C3 on C4 and retrolithesis of C6 on C7 and multilevel degenerative disc protrusions. Note the rather narrowed vertebral canal at C2/C3 secondary to posterior disc protrusion and ligamentum flavum hypertrophy and susceptibility artefact from surgical rod in situ between T1 and T2. (B) Ax T1w image at T1 level showing mild fatty atrophy within posterior cervical paraspinal musculature (arrow) particularly affecting the left Semispinalis coli of the transversospinales muscle group. (C) Ax T1w image at the level of the mid to distal thigh with increased T1 signal in the muscle groups of Semimembranosus muscle bellies (worse on the right) indicated fatty infiltration (arrowed). There is also mild fatty atrophy within the left biceps femoris muscle (asterisk). The rest of the thigh musculature is of normal volume. There is marked bilateral oedema and mild fatty atrophy within both tibialis anterior muscle bellies worse on the right (not shown). (D) STIR images at level at midthigh level with arrow indicating muscle oedema.

(A) This H&E-stained section (10×) of right vastus lateralis muscle biopsy shows increased variation in fibre size with no evidence of necrosis or regeneration. There is focal endomysial chronic inflammation. (B) This NADH-TR stained section (20×) of right vastus lateralis muscle biopsy shows presence of cores within muscle fibres. (C) This H&E-stained section (10×) of cervical paraspinal muscle biopsy shows increased variation in fibre size with several atrophic fibres and a few hypertrophic fibres, nuclear internalisation and focal perimysial chronic inflammation. (D) This NADH-TR-stained section (20×) of cervical paraspinal muscle biopsy shows presence of cores within muscle fibres.
Differential diagnosis
Axial weakness can be seen in myopathies, myasthenic and motor nerve disorders. Common metabolic causes of myopathy such as thyroid and parathyroid disorders were excluded with normal blood tests. Pompe disease which is a treatable hereditary cause was excluded biochemically through enzyme analysis. No antibodies against AChR and MuSK and the absence of jitter on single fibre EMG excluded a diagnosis of myasthenia gravis. Routine EMG confirmed myopathic changes. No structural spinal cord pathology was identified on MRI cervical spinal imaging but changes in the axial musculature were found as outlined above. We undertook a muscle biopsy of both paraspinal and quadriceps with the histological changes described above seen more prominently in the paraspinal rather than the quadriceps biopsy. The targeted muscle biopsy, along with clinical syndrome, helped establish the diagnosis of an axial myopathy.
Treatment
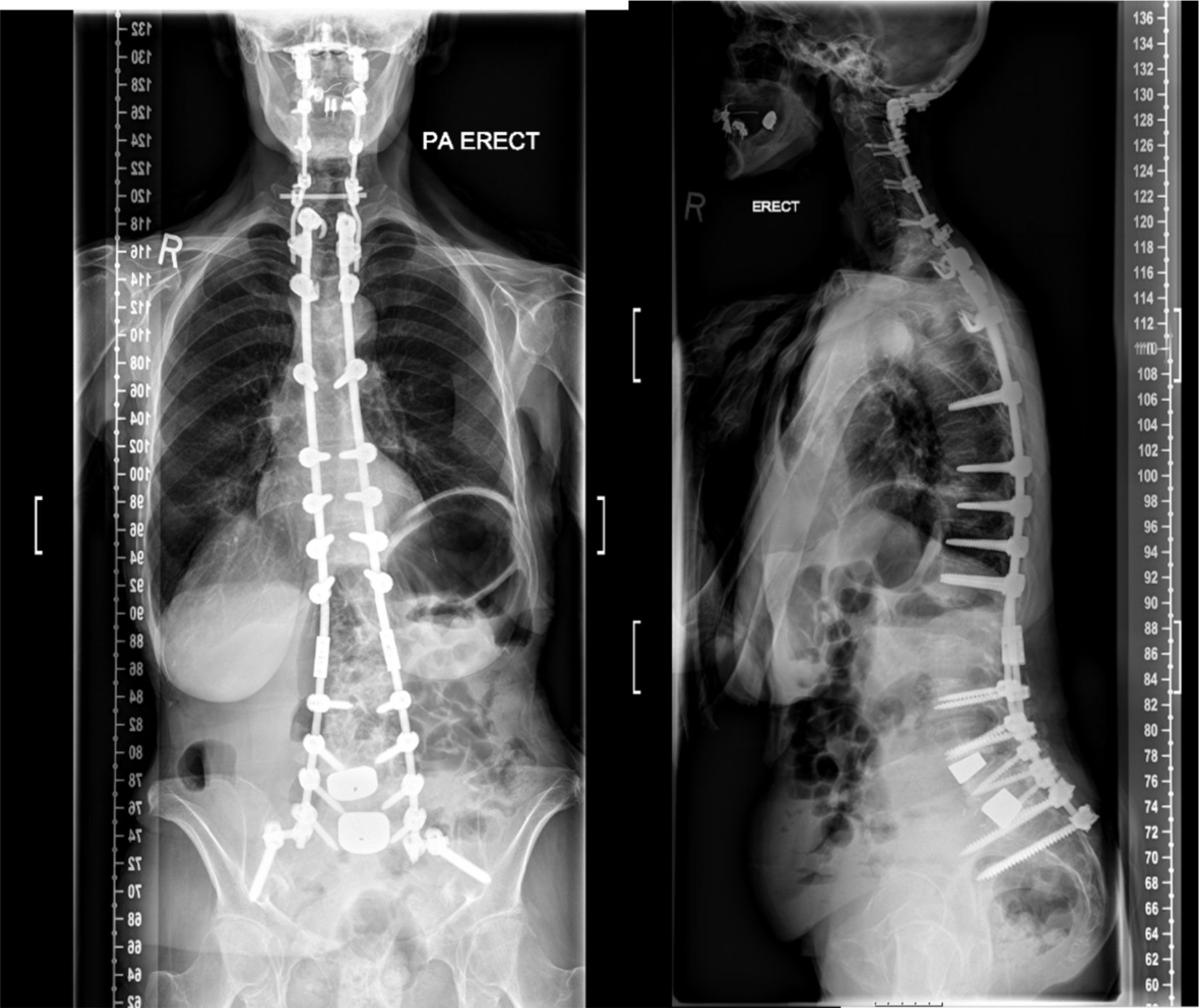
Axial myopathies can be difficult to treat. While there was minimal evidence of an inflammatory aetiology, our patient was very keen for a therapeutic trial of corticosteroid treatment but an 8-week trial led to no improvement in neck extensor strength. Over the next few months, the head drop worsened impeding her swallowing and various neck orthotic devices were tried without success. The patient was keen for further surgery and the dorsal fusion was extended into her neck (figure 3).

{kind=link}
{kind=link}
{kind=link}
Posteroanterior and lateral erect radiographs demonstrating surgical rod position. Note the subsequent new cervical fixation with occipital plate and mechanical connection to pre-existing thoracic and lumbar rods.
Outcome and follow-up
Although she is now unable to flex her neck, she reports a dramatic improvement in her quality of life and is able to resume her daily activities. Over the forthcoming years her myopathy has progressed slowly and she has also developed mild idiopathic Parkinson’s disease that has been treated conventionally with levodopa. She remains able to mobilise independently but now uses a stick for confidence purposes. Her lung function tests are monitored annually and remain satisfactory.
Discussion
Spinal deformity with axial myopathies can cause significant compromise to activities of daily living and quality of life. This case highlights that surgical treatment is a potential option for certain patients. Surgical intervention can have complications including new neurological deficits causing spinal cord and peripheral nerve injury as well as the common medical complications that occur after spinal surgery. Patients may also require significant postoperative rehabilitation. Published literature on complications related to adult spinal deformity surgery is mainly limited to scoliosis correction.2 A recent US study looking at over 33 000 patients aged 18–80 years who underwent posterior cervical fusion over a 12-year period found the rate of perioperative neurological deficit, morbidity and mortality were 1.08%, 40.44% and 1%, respectively.3 The predictors of surgical morbidity were increased age, cardiac disease, diabetes and pre-existing significant neurological deficits.
It is important to note that there is limited data available with regards diagnostic utility of paraspinal muscle biopsies as they are sampled rarely. In our institute, we routinely perform a lower limb (typically quadriceps) muscle biopsy as a control.
While captocormia and axial muscle weakness are phenomena described with Parkinson’s disease, at the time of initial presentation and for many years afterwards, there were no clear extrapyramidal signs or early symptoms such as anosmia or Rapid eye movement (REM) sleep behaviour disorder.
We could not find any dedicated studies related to spinal fusion surgery in the management of axial myopathies, although it has been used in cases of head drop secondary to cervical spondylosis.4 It is a fair assumption that surgery will carry a risk for new neurological deficits in addition to other complications. With respect to neuromuscular disorders, most published data are related to scoliosis correction surgery in Duchenne Muscular Dystrophy. These patients are of course much younger than our patient at the time of surgery, but will likely have significant cardiac dysfunction and respiratory muscle weakness. Surgical management of captocormia in Parkinson’s disease is reported although a meta-analysis identified high complication rates with spinal surgery.5
In our opinion, surgical intervention could be considered in selected axial myopathy cases where conservative and medical treatments have provided little benefit. Patients should be made aware of potential surgical complications and slow postoperative recovery. Also despite patient enthusiasm, coexisting comorbidities may preclude surgery, particularly in older patients.
Learning points
Use of MRI can complement clinical judgement in the diagnosis of an axial myopathy. Paraspinal muscle biopsies are sampled less frequently than other skeletal muscles and may be difficult to interpret.
Surgical intervention could be considered in cases where conservative and medical management have failed.
Surgery can lead to new neurological deficits and there may be a long postoperative recovery.
Acknowledgments
The authors would like to acknowledge Dr Dimitri Amiras (Consultant Radiologist) and Dr Clara Limback-Stanic (Consultant Neuropathologist) for providing images and assisting with the legends for the figures.
Footnotes
Twitter @charliewade
Contributors KK and SV helped in conceptualisation. KK performed the original draft preparation. CLF helped in figures. SV and JL helped in reviewing and editing.The authors would like to acknowledge Dr Dimitri Amiras (consultant radiologist) and Dr Clara Limback-Stanic (consultant neuropathologist) for providing images and assisting with the legends for the figures.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Patient consent for publication Obtained.
Provenance and peer review Not commissioned; externally peer reviewed.