Article Text

Statistics from Altmetric.com
Description
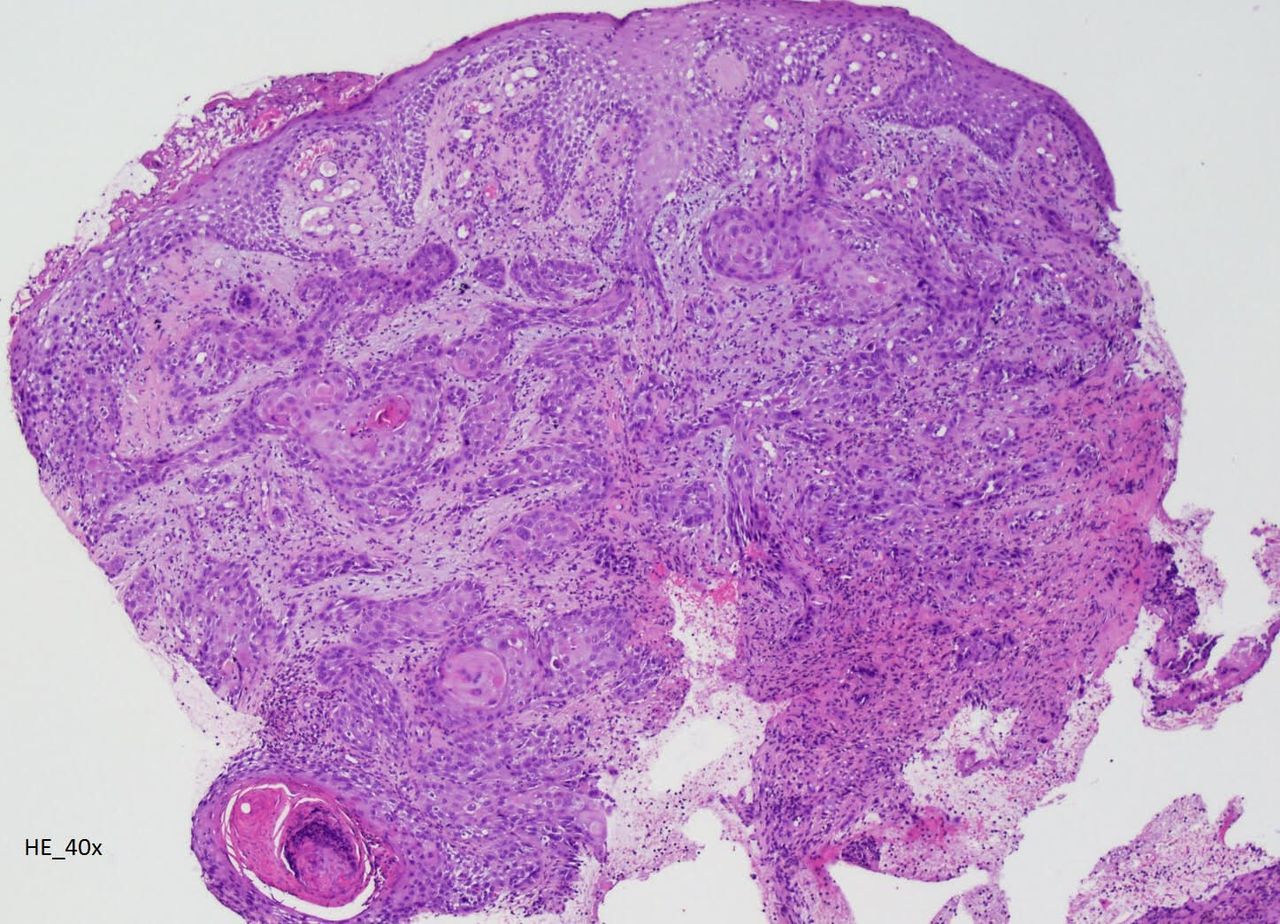
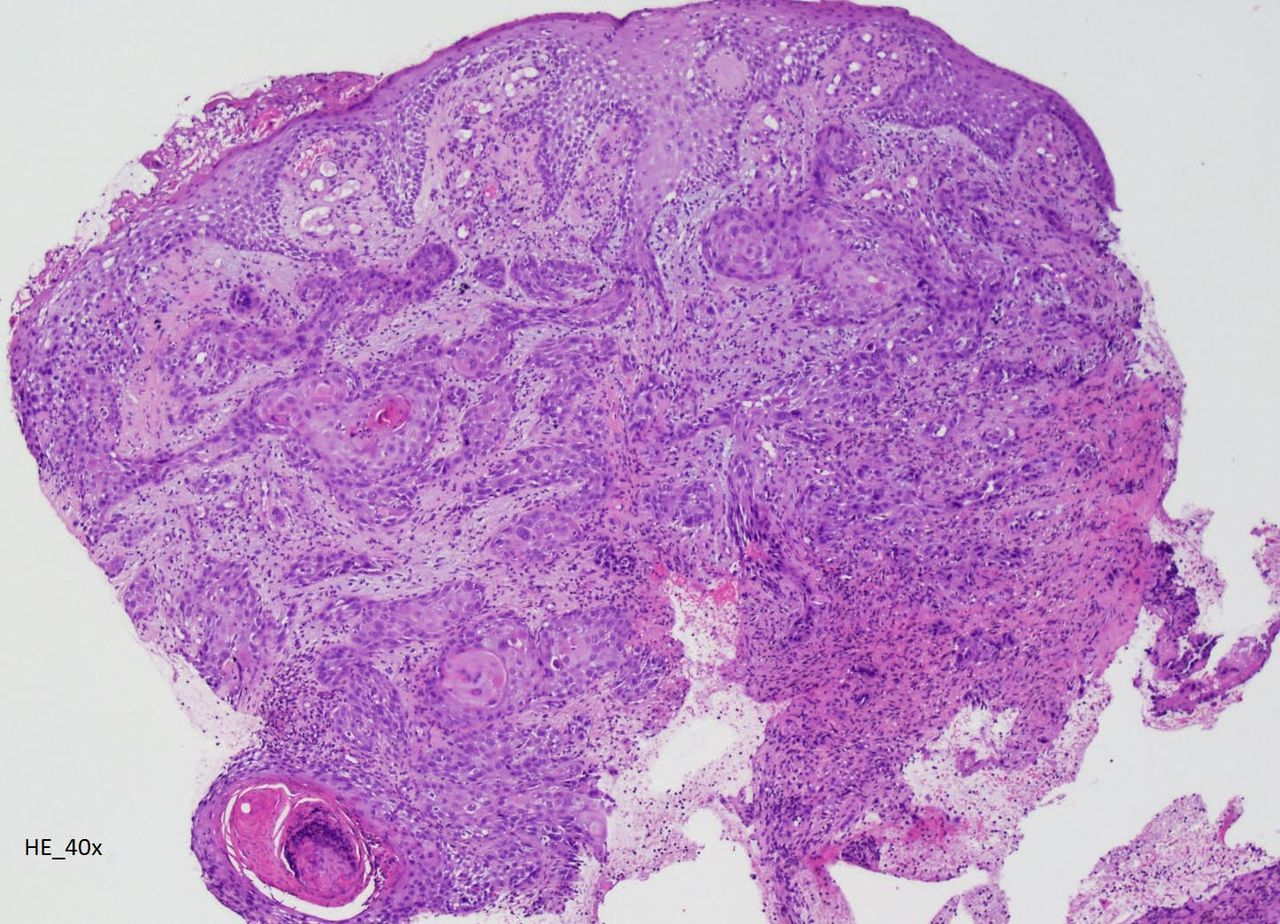
A 32-year-old man with congenital X-linked agammaglobulinaemia (XLA) immunodeficiency, receiving weekly intravenous immunoglobulin since childhood, was admitted for painful and ulcerative proliferative lesion with purulent exudate in the right ankle, with 1 month of evolution (figure 1), associated with walking disability and fever. Laboratory showed leukocytosis and elevated C reactive protein. Microbiology identified Morganella morganii and Pseudomonas aeruginosa. CT scan and MRI revealed an extensive infiltrative lesion in the lateral lower third of the leg and ankle, causing bone erosion of the peroneum and surrounding the peroneal, extensor and Achilles tendons. Lesion biopsy showed an extensive involvement by neoplasia with characteristics of invasive squamous cell carcinoma (figure 2). He was treated with piperacillin/tazobactam plus vancomycin before undergoing leg amputation.

Extensive ulcerative infiltrative lesion with purulent exudate in the lateral lower third of the right leg and ankle.
{kind=link}
{kind=link}
Lesion biopsy showing an extensive involvement by neoplasia with characteristics of invasive squamous cell carcinoma.
Primary immunodeficiency disorders (PIDs) result from defects in immune system development or function. PIDs are broadly classified as disorders of adaptive immunity (T-cell, B-cell or combined immunodeficiencies) or innate immunity (phagocyte and complement disorders).
XLA (also known as Bruton’s agammaglobulinaemia) is a PID characterised by lack or very low levels of immunoglobulins. It is a rare condition, accounting for 1 in 200 000 live births, that predominantly affects males.1 XLA is caused by mutations in the Bruton’s tyrosine kinase (BTK) gene, being inherited in an X-linked recessive manner.2 Other mutations have been reported, but all genes code for proteins that work with BTK to support the maturation of B-lymphocyte precursors into B-lymphocytes.3 People affected by this condition generally develop recurrent bacterial infections from about 6 months of age, like pneumonia, ear and skin infections. When suspected, agammaglobulinaemia should be screened, evaluating serum immunoglobulins and the number of B-cells in the peripheral blood (both are markedly reduced or absent in most patients with XLA). Confirmation of the diagnosis requires the detection of BTK mutation in DNA or the absence of BTK protein in monocytes or platelets. After infection, malignancy is the most prevalent cause of death in patients with congenital immunodeficiency disorders.4 Reports of tumours arising in patients with PID have been increasing in recent decades. This may be due to increased survival rate of these patients because of standard use of intravenous gammaglobulin (the main treatment for people with XLA) and a better management of recurrent infections. Different reports quote a 1.5%–2% risk of malignancy development among patients with XLA.5 It is notable that a relatively limited repertoire of cancers has been observed, mostly non-Hodgkin lymphomas and gastrointestinal cancers. However, literature reports about cancer incidence in XLA are conflicting, raising uncertainty about the cancer risk and localisation.6 7
Our patient thought the skin lesion was another infected ulcer, delaying healthcare-seeking. For the first time in the literature, a localised skin invasive squamous cell carcinoma has been diagnosed in a patient with XLA.
Lower serum immunoglobulin levels are associated with infections and malignancies, highlighting the importance of an effective immune response against infection and oncogenesis. Although lymphoproliferative disorders are the most prevalent, other malignancies can occur. Clinicians should be aware of the diagnosis of XLA and its complications.
Learning points
X-linked agammaglobulinaemia (XLA) is a rare primary immunodeficiency characterised by very low levels of immunoglobulins.
Patients affected with XLA experience recurrent and severe bacterial infections.
Reports of tumours are arising in patients with XLA, probably a consequence of the increased survival of these patients due to recent improvements in therapeutic strategies.
Ethics statements
Patient consent for publication
Footnotes
Contributors AS is the first author of the article. EC, VP and NO analysed and corrected the article.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.