Article Text

Statistics from Altmetric.com
Description
A 49-year-old woman was referred to the retina clinic with incidentally noted retinal pigmentation. She had long-standing poor left visual acuity (VA) due to a compressive optic neuropathy treated by surgery in childhood. She also had chronic angle closure glaucoma, treated with bilateral laser peripheral iridotomies (PIs) and left lensectomy. She was otherwise healthy with no active other medical conditions.
On examination, VA was 6/9.5 and 6/24 in right and left eyes, respectively. She had a left relative afferent pupillary defect, patent bilateral PIs, left pseudophakia and intraocular pressure of 12 mm Hg bilaterally. There was no evidence of intraocular inflammation. While the right optic disc was normal, the left eye had significant cupping and pallor. There was symmetrical perivascular nummular pigmentation with associated chorioretinal atrophy most notable at the superotemporal vascular arcades (figure 1A,B). Autofluorescence imaging highlighted the extent of retinal pigment epithelial (RPE) atrophy in the same distribution, with less marked mottling of RPE autofluorescence nasally (figure 1C,D). Optical coherence tomography showed preserved foveal photoreceptor structures, no macular oedema/schisis and absent retinal nerve fibre layer in the left eye, in keeping with advanced optic neuropathy. Static perimetry showed advanced visual field loss in the left eye likely due to the advanced optic neuropathy and an early ring scotoma in the right eye more marked inferiorly in keeping with the distribution of retinal atrophy (figure 2A,B). Rod responses were absent on full field electroretinogram and cone responses were delayed and attenuated with the pathognomonic bright flash a-wave amplitude greater than 30 Hz flicker amplitude. Panel-based next-generation sequencing (NGS) detected a homozygous likely pathogenic mutation (c.305C>A,p.Ala102Asp) in the NR2E3 gene (15q23) which has been reported as responsible for autosomal recessive enhanced S-cone syndrome (ESCS), also known as Goldmann-Favre syndrome.

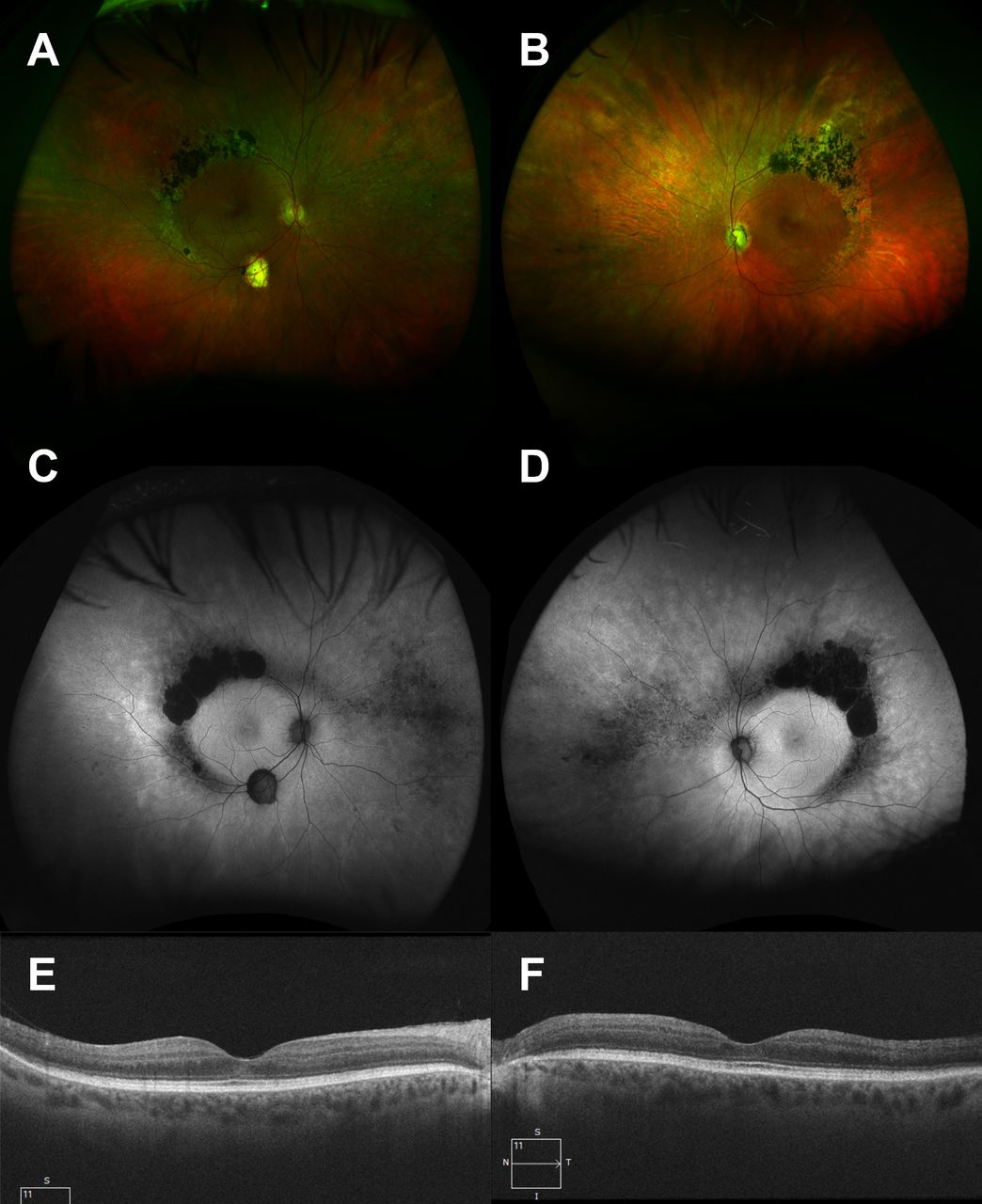
Widefield colour Photographs (T200x camera, Optos plc, UK) of the right (A) and left (B) eyes showing predominantly superotemporal perivascular nummular pigmentation and outer retinal atrophy/pallor. There are subretinal white dots and less marked intraretinal pigment hypertrophy nasally. The left optic disc shows cupping and pallor and there is an isolated area of retinal pigment epithelial (RPE) atrophy at the right inferotemporal vascular arcade. Widefield autofluorescence imaging (T200x camera, Optos plc, UK) of right (C) and left (D) eyes delineates the extent of RPE atrophy, revealing hyperautofluorescent areas peripherally throughout the fundus with preserved macular isoautoflourescence. Optical coherence tomography (Cirrus 5000, Carl Zeiss MediTec, Dublin, California, USA) shows preservation of the photoreceptor layers within the macula (E, F).
{kind=link}
{kind=link}
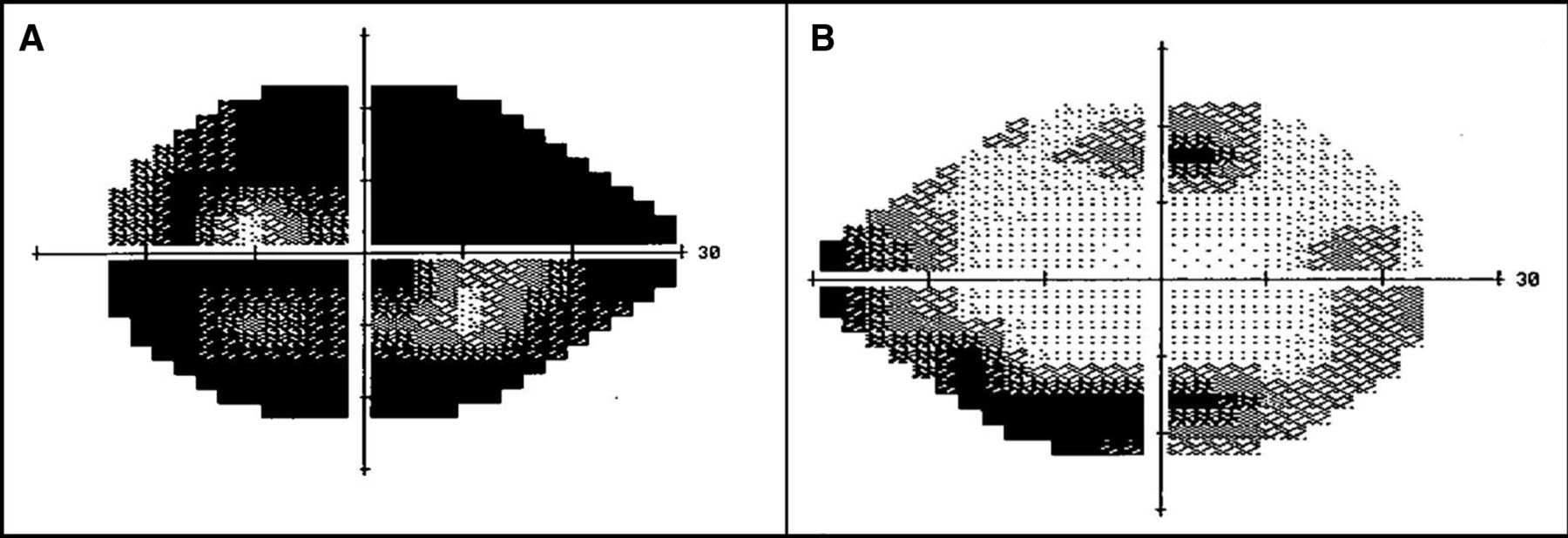
Static visual fields (24–2, Humphrey field analyzer, Carl Zeiss MediTec, Dublin, California, USA) showing advanced reduction in sensitivity of left eye (A) due to multifactorial optic neuropathy, while the right eye (B) has an incomplete ring scotoma corresponding to the outer retinal atrophy at the vascular arcades.
Confirmation of genotype is relevant as novel gene therapies are under investigation for multiple inherited retinal degenerations (IRDs); panel-based NGS investigation genetically resolves IRD pedigrees in >70%.1 2 ESCS is a disorder of photoreceptor differentiation in which short-wavelength sensitive cone photoreceptor (S-cone) progenitors fail to fully differentiate into rods or long/medium wavelength-sensitive cone photoreceptors3 supported by pathological (double normal cone number,~12 million per eye, with 92% staining for S-cone markers)4 and electrophysiological (absent rod responses and 30 Hz amplitudes less than photopic bright flash a-wave)5 evidence. ESCS is caused by mutations in the NR2E3 gene (or rarely the NRL gene6) with 53 known pathogenic or likely pathogenic variants reported. Most NR2E3 variants cause ESCS while a single variant (c.356G>A,p.Gly56Arg) manifests as an autosomal dominant retinitis pigmentosa phenotype.7 8 ESCS is rare, with less than 1000 cases reported in the literature. Symptoms are typically early night blindness (~4 years), but this may be masked by other disease.5 At a mean of 26.6 years, VA was ≥6/12 in 57% of eyes, with 66% of patients meeting VA standards for driving and only 7% considered legally blind.5 Gradual progression (six letter loss over 6 years) is reported in only 21%5 and retention of visual field has been demonstrated.9 There is no approved genetic therapy to date; however, confirmation of an NR2E3 genotype aids prognostication.
Learning points
Primary photoreceptor disease may be masked by other pathologies, whether systemic (eg, intellectual disability in syndromic inherited retinal degeneration (IRD)) or ocular (eg, optic neuropathy).
Autofluorescence imaging is invaluable in assessing the health of the retinal pigment epithelium, helping to delineate symmetry and extent/severity of disease
Although therapies are not yet available/approved for most IRDs, some conditions are entirely/largely stationary and may benefit from genetic diagnosis for prognostic and family planning, and may allow access to future gene therapy.
Ethics statements
Patient consent for publication
Acknowledgments
Fighting Blindness Ireland. We thank the patient for consenting to share their case, understanding that their story could provide valuable insight for other healthcare practitioners in the field.
Footnotes
Twitter @KarkStaphonsen, @docdockgoose
Presented at Royal Academy of Medicine in Ireland, Ophthalmology Division, Winter Meeting 2018.
Contributors KGS contributed to the manuscript construction as well as aspects of the literature review. KAJS handled patient care, obtained consent, obtained imaging and approached me about writing up this case. AD was in charge of the genetic analysis and DK oversaw the entire project. All authors contributed to editing and formatting of the final manuscript.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.