Article Text

Statistics from Altmetric.com
Description
Cardiomyopathies (CMs) are myocardial disorders that lead to compromised cardiac function of variable severity. These diseases are very rare in human fetuses. Many CMs are idiopathic and only 33%–43% have identifiable genetic, familial, infectious or metabolic causes.1 2 About 76% of the cases of hypertrophic CM (HCM) are related to maternal diabetes and twin-to-twin transfusion syndrome. CM related to these conditions is often reversible.2
HCM caused by familial genetic variants is rare but leads to low fetal survival2 or serious long-term clinical consequences including a higher risk of sudden cardiac death.3 4 Prenatal diagnosis of HCM is uncommon and challenging with a reported incidence of 6.2 per 100 000.2 To the best of our knowledge, only isolated case reports and small case series of HCM diagnosed during fetal life have been reported.2
A 32-year-old healthy woman, gesta 5 para 1 (1 healthy child and 3 previous first trimester spontaneous miscarriages), with no gestational diabetes was referred to our prenatal diagnosis unit at 23 weeks of gestation due to a familial history of HCM diagnosed during the present pregnancy. The genetic study of the affected family member (mother of the pregnant woman diagnosed with HCM at 53 years of age) detected the variant c.559A>G (p.Asn187Asp) in heterozygosity for the MYH7 gene (NM_0002547.4). The presence of the family variant in the pregnant women was unknown and her echocardiogram was found to be normal. The fetal echocardiogram (at 23 weeks) showed a symmetric thickening of the fetal biventricular cardiac walls and interventricular septum, measuring about 5 mm and 4.5 mm, respectively. A preserved cardiac function, normal venous and arterial Doppler, and no obstacle in the ventricular outflow tract were observed (figure 1). The remaining fetal morphology was normal. Fetal karyotype was normal. The family variant in heterozygosity for the MYH7 gene was detected in the fetus by DNA sequencing. The patient decided for termination of pregnancy at 24 weeks of gestation.

The fetal echocardiogram (23 weeks) revealed normal heart size, symmetric thickening of the biventricular walls and interventricular septum, measuring about 5 mm and 4.5 mm, respectively.
The autopsy confirmed a fetus with a 20-week biometry, with a heart with normal weight but concentric biventricular hypertrophy (figure 2) and no other morphological anomalies.

{kind=link}
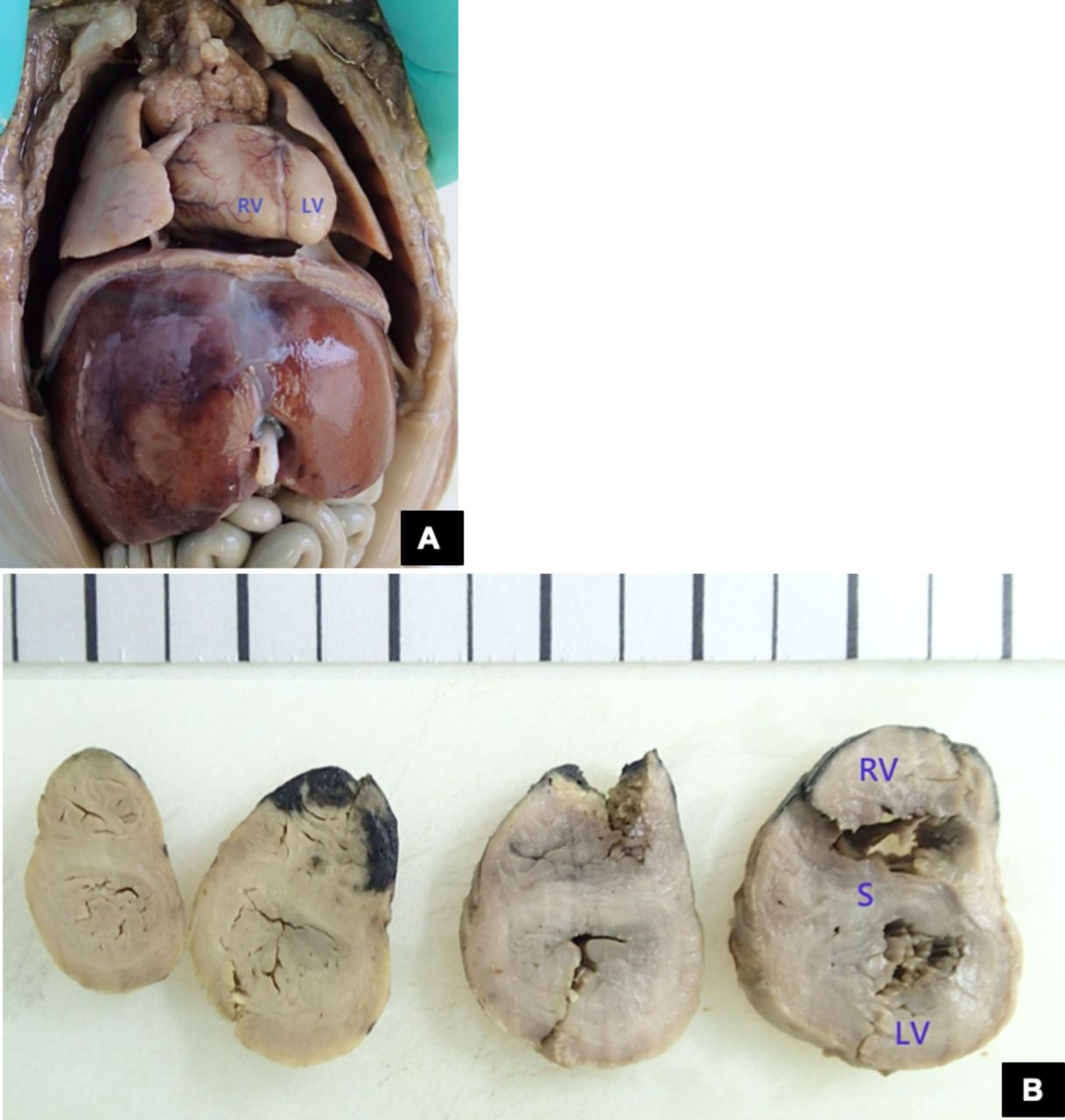{kind=link}
(A) In situ view of the heart. (B) Serial section showing biventricular hypertrophy and septal hypertrophy. Right ventricular (RV) thickness 5 mm; left ventricular (LV) thickness 4 mm; septal (S) thickness 3–5 mm. Courtesy of CGC Genetics, Embryofetal Pathology Laboratory.
In the fetus, a detailed diagnosis of the type of CM is very difficult, and only an initial assessment of these disorders based on the sonographic appearance of the myocardium is feasible. Furthermore, prenatal diagnosis of HCM is challenging, especially at an early stage, due to its highly variable expression and unpredictable evolution.1–3
In this report, an HCM case was diagnosed at 23 weeks of gestation, which is in line with the few studies about this condition.2 It should be noted that some authors reported normal scans at 20 weeks5 or just detected HCM at 30+ weeks.3 Timely HCM diagnosis and accurate prediction of functional outcomes are of paramount importance for pregnancy management, especially when a termination is an option and legal time limits must be respected (eg, 24 weeks in Portugal).
In the presented case, a cardiac morphological alteration comprising ventricular thickening with a normal function was observed, which is in line with one of the most important studies about HCM.2 Due to a recent finding of a familial history of CM with a known genetic variant, a targeted aetiological assessment was carried out.
A key finding presented in this report is the variant MYH7 (NM_000257.4):c.559A>G (p.Asn187Asp), which is described for the first time and is likely pathogenic. This specific variant was not described in healthy people in gnomAD v3.1, and the predictive software favoured the classification as pathogenic.6 7
Gene testing for cardiac β‐myosin heavy chain gene (MYH7) variant has been used in the clinical evaluation of fetal CMs.2 8 In fact, pathogenic variants of MYH7 have been found prenatally in non-compaction CMs6 and dilated CMs.2 However, there is just a single case report of HCM diagnosed prenatally pointing to an MYH7 variant as the cause5 but involving a distinct MYH7 variant than reported here.
Fetal CM prognosis is reported as extremely poor with a high risk of perinatal demise.2 The severity of HCM appears to be closely associated with the phenotypical and functional presentation, and more adverse perinatal outcomes are observed in cases with early echocardiographic findings.2
Learning points
In the fetus, only an initial assessment of cardiomyopathy (CM), based on the sonographic appearance of the myocardium, is feasible.
Prenatal diagnosis of hypertrophic CM (HCM) is rare. Due to its highly variable expression and unpredictable evolution, it is challenging to obtain a timely CM diagnosis during pregnancy.
MYH7 variant may be the cause of HCM diagnosed prenatally.
Ethics statements
Patient consent for publication
Footnotes
Contributors CS wrote the manuscript and acquired the data. RNN acquired the data and provided critical revision of the manuscript. CL and AC provided critical revision of the manuscript.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.