Article Text

Abstract
The CHEK2 gene is mostly considered as a moderate breast cancer gene with the result that many clinicians have a narrow focus. We present the 10-year journey of a man who had five different cancers and had iterative genetic testing including for Li-Fraumeni syndrome, eventually to discover a pathogenic variant in the CHEK2 gene, possibly explaining his numerous cancers. This diagnosis offered him closure which he had desperately sought for well over a decade. A pathogenic variant in the CHEK2 gene can potentially explain these cancers because of its function as a tumour suppressor gene. Consideration is warranted of what this means for individuals with CHEK2 variants who may develop multiple cancers, their prognosis and whether different treatment modalities such as chemotherapy, radiotherapy or target agents would need modification. We encourage more research into the many faces of the CHEK2 gene and the potential for predisposition to multiple cancers.
- genetics
- breast cancer
- endocrine cancer
- radiotherapy
- urological cancer
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
The CHEK2 gene is well researched and published as a moderate breast cancer risk gene.1 The frequency of the CHEK2*1100delC allele is 0.3% in North America and lower than the respective frequency observed in European populations.2
Clarity about the importance of CHEK2, in the predisposition towards other cancers, has been gained slowly. CHEK2 is a cell cycle checkpoint regulator and a tumour suppressor. The CHEK2 gene is activated by phosphorylation of Thr68 by ATM, which causes the dimerisation of the gene enabling it to acquire kinase activity. CHEK2 then reacts with downstream phosphatase CDC25, serine/threonine protein kinase NEK6, transcription factor FOXM1, p53 protein and BRCA1 or BRCA2.3 CHEK2 regulates cell division by preventing cells from entering mitosis or arresting cell cycle in gap 1 phase (G1), in response to DNA damage.4 Furthermore, CHEK2 protein interacts with several other proteins including p53 and stabilisation of p53 by CHEK2 leads to cell cycle arrest in G1.5 There are multiple variants recognised within CHEK2 such as 1100delC, I157T, R117G, I160M, G167R and G167A.6 The 1100delC variant is the most studied and implicated with breast and other cancers.7
CHEK2 mutation and breast cancer risk are well studied. According to Cybulski et al, the OR was higher for women with a first-degree or second-degree relative with breast cancer (OR 5.0; 95% CI 3.3–7.6) than for women with no family history (OR 3.3; 95% CI 2.3–4.7).8 CHEK2 in linked with multiple cancers including prostate cancer: 1100delC mutation (OR 3.29; 95% CI 1.85–5.85; p=0.00) and I157T missense mutation (OR 1.80; 95% CI 1.51–2.14; p=0.00).9 The age-adjusted and sex-adjusted HRs were 5.76 (95% CI 2.12–15.6) for stomach cancer, 3.61 (95% CI 1.33–9.79) for kidney cancer and 3.45 (95% CI 1.09–10.9) for sarcoma,10 and in a meta-analysis of a total of six studies including 4194 cases and 10,010 controls, a significant association of the CHEK2 1100delC variant with unselected colorectal cancer was found (OR 2.11; 95% CI 1.41-3.16; p=0.0003).11
A decade ago, CHEK2 was considered as predisposing to a possible ‘Li-Fraumeni-like’ phenotype12 because of its role as a Tumour suppressor gene.13 14 When considered together with the family history of young-onset breast cancer and multiple tumours including glioma, this case is clinically suggestive of a Li-Fraumeni-like syndrome.
Case presentation
A 52-year-old man was diagnosed with five cancers; germline DNA testing detected the heterozygous pathogenic variant, CHEK2 1100delC.
Clear cell renal cancer
In 2007, the patient, aged 40 years, was found to have 2 cm renal cell cancer in the right kidney, following an ultrasound scan for urinary tract symptoms. Laparoscopic right nephrectomy was done, and pathology was consistent with clear cell carcinoma of Furman grade 2. There was no perinephric or adrenal gland involvement.
The immunohistochemistry (IHC) examination of the tumour for succinate dehydrogenase B (SDHB) showed a normal pattern of staining, indicating that it was unlikely to harbour SDHx gene mutation.
His maternal aunt was concurrently diagnosed with pheochromocytoma at the age of 56 years, and the c.74C>T(p.pro25Leu) variant in the VHL gene was detected in her DNA, regarded at that time as pathogenic.
Glioma
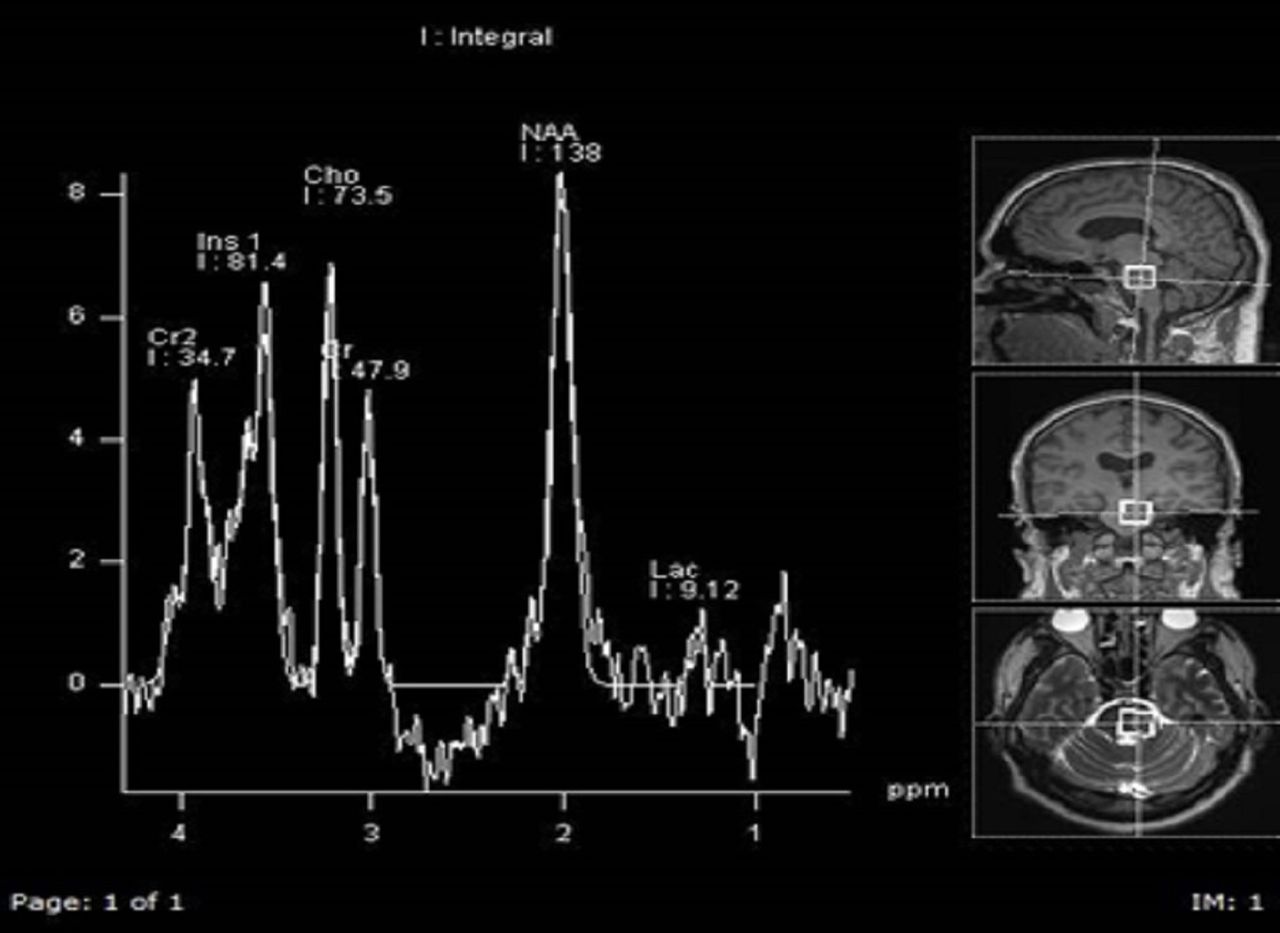
In 2010, as part of the investigation of a possible diagnosis of von Hippel-Lindau syndrome, a brain MRI was done and demonstrated a possible low-grade glioma/astrocytoma in the right temporal lobe and diffuse signal abnormality in the left cerebellum and left temporal lobe. Brain MRI and the clinical picture was suggestive of the possibility of multifocal astrocytoma or a low-grade glioma. Figure 1 shows these features on the MRI scan. Figure 2 shows spectroscopy finding of increase choline that is compatible with a low-grade glial tumour. No biopsy was done as it was deemed too much of a risk at the time. The tumour was observed over time and treatment with radiotherapy was initiated in 2013 (50.4G/28#) as there were progressive symptoms with diplopia and dysarthria. Radiotherapy and a trial of steroid did not alleviate his symptoms, although his 6 monthly MRIs have remained unchanged.

MRI.
{kind=link}
{kind=link}
MR spectroscopy.
Low-grade urothelial tumour
In 2011, a low-grade urothelial tumour was detected in his postnephrectomy follow-up. He had persistent lower urinary tract symptoms; cystoscopy detected a tumour adjacent to the left ureteric outlet, consistent with low amplitude papillary urothelial cancer. This was surgically managed, and there is no evidence of recurrence to date.
Cutaneous basal cell cancer
In 2016, he had a non-healing ulcer in his forehead, which was a basal cell carcinoma. This was surgically resected with no postoperative complications.
Multifocal bilateral papillary thyroid cancer
In 2019, he was diagnosed with bilateral multifocal papillary thyroid cancer that was breaching the capsule on the left side, but with no infiltration of overlying muscle. He had a completion thyroidectomy on the contralateral side, which detected an additional 5 mm papillary thyroid cancer. He had radioiodine therapy and is currently on thyroxine replacement with no evidence of metastasis.
Family history
His mother died of non-Hodgkin’s lymphoma, positive for B-cell markers at the age of 47 years, in 1993. Her maternal aunt had pheochromocytoma at the age of 56 years and breast cancer at 66 years. IHC for SDHB was preserved in her pheochromocytoma specimen. The proband’s sister was diagnosed with grade 3 estrogen positive (ER+) breast cancer at the age of 26 years and died at the age of 27 years, in 1999. Neither his sister nor mother had genetic testing.
Genetic testing
The proband’s maternal aunt, who lived a significant distance away had genetic testing; no pathogenic mutation was detected in either BRCA 1 or BRCA2 genes. A variant identified in the VHL gene c.74C>T(p.Pro25Leu) was thought to be pathogenic at the time, and the extended family was advised that family testing was available. On this basis, our proband was offered testing for the family-specific variant. The DNA sample obtained from the patient was independently amplified and sequenced in opposite directions from exon 1 of the VHL gene. Analysis of this exon has not detected the c.74C>T variant. Soon thereafter, this variant was revised as unlikely to be pathogenic.
Subsequently, his aunt had next-generation sequencing (NGS) for MAX, RET, SDHAF2, SDHB, SDHC, SDHD, TMEM127 and multiplex ligation-dependent probe amplification (MLPA) for VHL, SDHA, MAX, SDHB, SDHC and SDHC. No pathogenic mutation was detected.
Our proband was offered MLPA and NGS for VHL and TP53. Complete sequence screening of all coding exons and intron–exon boundaries of the TP53 gene did not reveal the presence of a pathogenic variant. In MLPA, none of the tested exons showed a significant deviation from the normal range of the tested population, and duplicate tests were concordantly indicating a normal copy number.
Following his disease progression and with the recent diagnosis of papillary thyroid cancer as well as papillary urothelial cancer and the availability of broader panel testing, testing of72 cancer predisposition genes was offered for him. The heterozygous pathogenic variant, CHEK21100delC was found.
Outcome and follow-up
Confirmation of a pathogenic mutation in the CHEK2 gene provided an answer for his propensity to cancers, affording him closure. The most recent CT scan done on the 13 May 2020 did not detect any disease recurrence of the thyroid and urothelial cancers. His MRI scans of the brain remain radiologically stable despite having subtle clinical progression occurring over a few years. We have offered predictive testing to his relatives; this is an autosomal dominant gene and therefore, his siblings and offspring would have a 50% chance of having the same variant. We were not able to test his aunt for the CHEK2 variant to date and verify the presence and variant segregation with the phenotypes. Genetic counselling will be provided to his extended family.
Discussion
This man meets the modified Chompret criteria15 with multiple tumours including glioma, clear cell renal cell cancer, urothelial cancer, basal cell cancer as well as multifocal bilateral papillary thyroid cancers in combination with the family history. However, only cancer this man suffered from which is described in the Li-Fraumeni syndrome spectrum of cancers is a glioma. In the absence of a pathogenic variant in the TP53 gene, we posit that his cancers are driven by the CHEK2 mutation, potentially behaving like TP53.
Inherited RCC comprises 5% of all renal cell cancers,16 based on germline mutations in the VHL, FH, BAP1, SDHB, SDHC, SDHS, TSC1, TSC2 and MITF17 genes. CHEK2 has been implicated in RCC, especially the missense variant I157T.18 However, literature around CHEK2 1100delC is less clear. According to Näslund-Koch et al,10 CHEK2*1100delC HR for kidney cancer is 3.45 (95% CI 1.09–10.9) that supports the theory of driving renal cell cancer, but CHEK2*1100delC was not observed in any of the patients with renal cell cancer in Huzano and Kolosza,19 therefore, the data supporting the role of CHEK2*1100delC are conflicting.10 17 19
The link between CHEK2 mutation and gliomas is plausible because of its role as a tumour suppressor gene; case reports published suggest CHEK2 mutation as the possible cause of medulloblastomas,20 as well as primary gliomas and astrocytomas.21 Germline mutations in CHEK2 do not seem to cause either high-grade or low-grade gliomas, in contrast to the TP53 gene.22 No biopsy was undertaken, so no tissue was available to seek somatic mutations.
Genotyping may be useful for the radiation oncologist when determining treatment options. The primary concerns include increased radiation toxicity, secondary cancers or lack of efficacy in certain tumours.
Truncating mutations in ATM causing breast cancer and radiation toxicity is one such instance.23 There are concerns about TP53 mutations and an increased risk of secondary cancer24 or lack of radiation sensitivity with germline mutations.25 There is insufficient information about CHEK2 mutation and radiation. Still, it is possible that the tumour had a degree of radioresistance as evident by the lack of response to treatment.
In terms of his development of basal cell cancer, we believe radiotherapy might have played a role as his basal cell cancer developed on his forehead. Indeed few publications are confirming such existence, but the majority of patients do not survive after radiotherapy to develop secondary cancers such as basal cell carcinoma.26 However, the role of CHEK2 mutation in itself driving this or increasing radiosensitisation as a result of the underlying mutation cannot be discounted as well. We hypothesise the development of his basal cell carcinoma in this male to be most likely multifactorial.
The link between thyroid cancer and CHEK2 is well published (OR 4.9; p=0.0006).18 In published data, CHEK2 accounted for more than 15% of mutations found in an unselected population. If women are selected who had thyroid cancer with a family history of breast cancer, it becomes even more significant (OR 10: p=0.0004).27
We hypothesise that the pathogenic variant in the CHEK2 gene has rendered him more susceptible to the multiple cancers he has developed. It is reasonable to consider CHEK2 as a gene with the potential to drive multiple cancers, comparable with Tp53, rather than merely a moderate breast cancer risk gene.
Another important new paradigm in genetics is the concept of polygenic risk scores. This is the concept of several low penetrant variants, further enhancing the lifetime risk of cancers and this well-described concerning CHEK2 pathogenic variant in breast cancer.28 Unfortunately, these models are still not incorporated at a clinical level, and we expect this to change in the future.
On the basis of this resurgence of support for a Li-Fraumeni-like phenotype and the importance of incorporating this into polygenic risk scores for risk management, we suggest more investigation of this gene.
Patient perspective
More than a decade ago my health journey started when I started going to the toilet at least five times a night, so the next day I made an appointment to see my doctor. He sent me for an MRI of my urinary tract which revealed a tumour on my right kidney which I had to get it removed. Then I had further scans of my urinary tract which revealed a tumour in my bladder. They took a biopsy, the result stated that it was cancer. I had an operation to remove it. My cousin told me that I should have genetic testing done because my mum passed away in her 40s due to non-Hodgkin lymphoma and my sister in her 20s due to breast cancer with a toddler.
A couple of months later I had a transurethral resection of the prostate (TURP). The MRI that I have every 6 months showed that I have glioma in my brainstem. I had undergone radiotherapy to my brain, which did not work. My symptoms did not change, my balance, vision and coordination have not improved. At my age, it will get worse. It is very frustrating. Unfortunately, the doctors cannot operate on the glioma because it is too dangerous. The MRI that I have every 6 months revealed that I have a growth within my thyroid, which I had half removed and 3 months later, had the other half removed. My ordeal carries on but I have not felt sorry for myself. I think positively and live my life to the best of my ability. Then my treating doctors found a gene connected to me after many years of research which is a big relief to me. Not knowing why I was getting so many cancers was very frustrating. Getting the news from my doctor brought a big relief to me and my family. I have a big road ahead of me which I will overcome.
Positive thoughts to one and all.
P.S. I would also like to thank Professor and all of the staff who cared for me at the tertiary hospital, also the professors, doctors, nurses and reception staff at my local cancer centre. My cancer centre treats patients with the utmost respect.
Learning points
CHEK2 mutations should be thought of as a gene mutation that can drive multiple cancers.
More studies are required to ascertain the treatment effects of chemotherapy and radiotherapy for patients who have lesser-known germline mutations.
Since the field of genomics is evolving rapidly, we need to keep searching for answers until we are satisfied all the questions have been answered.
Genomics can be an important tool in the armoury that can provide closure to cancer patients and restore confidence in health professionals.
Acknowledgments
University Of Melbourne Parkville Familial cancer Centre Victoria Australia.
References
Footnotes
Contributors DLDS contributed towards the planning, conception, design of the work, analysis and interpretation of data and the final production of the manuscript. DLDS have drafted the work and revisited it critically for important intellectual content. DLDS is accountable for the final approval of the version published. DLDS is also accountable for all aspects of the work in ensuring that questions related to the accuracy or integrity of any part of the work are appropriately investigated and resolved. IW contributed towards the planning and conception, design of the work, analysis, interpretation of data and supervision of DLDS.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Patient consent for publication Obtained.
Provenance and peer review Not commissioned; externally peer reviewed.