Article Text

Abstract
Histamine is involved in various physiological functions like sleep–wake cycle and stress regulation. The histamine N-methyltransferase (HNMT) enzyme is the only pathway for termination of histamine neurotransmission in the central nervous system. Experiments with HNMT knockout mice generated aggressive behaviours and dysregulation of sleep–wake cycles. Recently, seven members of two unrelated consanguineous families have been reported in whom two different missense HNMT mutations were identified. All showed severe intellectual disability, delayed speech development and mild regression from the age of 5 years without, however, any dysmorphisms or congenital abnormality. A diagnosis of mental retardation, autosomal recessive 51 was made. Here, we describe a severely mentally retarded adolescent male born from second cousins with a homozygous mutation in HNMT. His phenotypic profile comprised aggression, delayed speech, autism, sleep disturbances and gastro-intestinal problems. At early age, regression occurred. Treatment with hydroxyzine combined with a histamine-restricted diet resulted in significant general improvement.
- psychiatry (drugs and medicines)
- metabolic disorders
- genetics
- therapeutic indications
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
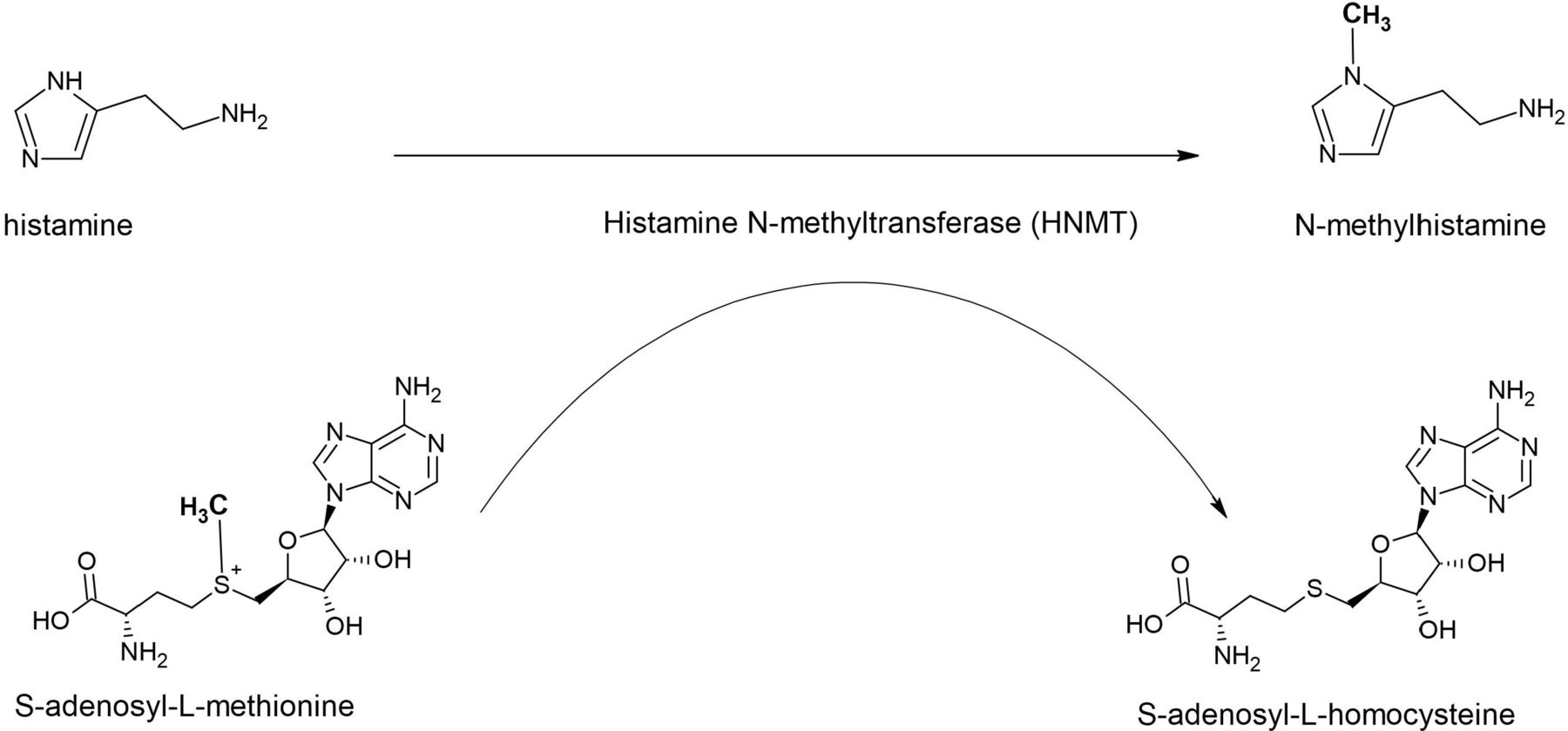
Brain histamine, formed from the essential amino acid L-histidine, is a neurotransmitter and involved in several physiological functions like sleep–wake cycles, stress response and appetite. Two different enzymes are responsible for its inactivation, that is, diamine oxidase (DAO) and histamine N-methyltransferase (HNMT), located in chromosome 2q22.1 (OMIM: 605238). DAO, also known as histaminase, is mainly expressed in the digestive tract and to a lesser extent in the kidneys and placenta, which indicates that this enzyme metabolises histamine in the peripheral organs but not in the central nervous system (CNS). In contrast, HNMT is widely expressed in the CNS, kidney and liver and catalyses the transfer of a methyl group from S-adenosyl-L-methionine to histamine, yielding N-methylhistamine and S-adenosyl-L-homocysteine (figure 1). Thus, inactivation of histamine by HNMT is the only well-known pathway for the termination of neurotransmission action in the mammalian CNS.

Enzymatic activity of histamine N-methyltransferase (HNMT). HNMT catalyses the transfer of a methyl group from S-adenosyl-L-methionine to histamine, yielding N-methylhistamine and S-adenosyl-L-homocysteine (figure provided by PKCJ).
As reported by several investigators, genetic single nucleotide polymorphisms of HNMT may play a role in a variety of human brain disorders such as Parkinson’s disease,1–3 and attention deficit disorder,4 although that it is still not elucidated whether alterations in HNMT are primarily or secondarily involved.5 In addition, lowered histamine levels in cerebrospinal fluid have repeatedly been reported in patients with narcolepsy and other disorders with excessive daytime sleepiness.6 7 Finally, it is not clear whether polymorphisms of HNMT are also associated with gastrointestinal diseases.8
Animal experiments with HNMT knockout (KO) mice have demonstrated that HNMT deficiency enhances brain histamine concentrations indeed. As a consequence, the histamine KO mice showed high aggressive behaviours and experienced dysregulation of sleep–wake cycles.9 10
In recent years, a very limited number of patients have been described with a genetically caused deficiency of HNMT. One patient, aged 23 years, with a deletion at 2q22.1q22.3 encompassing among others the HNMT gene showed a clinical picture characterised by severe intellectual disability and several somatic anomalies related to other deleted genes.11 Apart from this patient, two unrelated consanguineous families of Turkish and Kurdish ancestry, respectively, have been reported.12 The Turkish family with its background in Iraq had a total of nine children of whom four (two boys and two girls) showed profound to severe intellectual disability and speech was limited to single words. The condition was milder in the males as compared with the females and none of them had any neurological problems nor dysmorphisms, autism or congenital anomalies of any kind. All affected members showed mild regression from the age of 5 years. The Kurdish family with its origin in Iraq had seven children of whom three (two boys and one girl) presented with severe intellectual disability and delayed speech development. Similarly, these patients had no dysmorphisms or any congenital malformations. Exome sequencing identified two different homogeneous missense HNMT mutations. In both families with affected members, ranged in age from 13 years to 35 years, a diagnosis of non-syndromic autosomal intellectual disability was established that was named mental retardation, autosomal recessive 51 (MRT51; OMIM: 616739). These observations indicate that histamine modulates brain development and that HNMT plays an important role in human neurodevelopment.
Here, we describe in detail an adolescent male with severe intellectual disability from a family of second generation consanguinity in whom exome sequencing finally yielded a homogenous mutation in the HNMT gene.
Case presentation
Early development
The patient is a 23-year-old severely intellectually disabled native Dutch man born from consanguineous parents in that the paternal grandmother of the patients’ father was a sister of the paternal grandmother of the patient’s mother. Father and mother are, therefore, second cousins. The patient was born after his mother had five miscarriages and one healthy son. Pregnancy and delivery were uncomplicated and during his first 2 years, development of speech, motor function and sociality were normal, although that he suffered from recurrent upper airway infections. Thereafter, however, global deterioration with loss of previously acquired capacities, especially language, became apparent and severe aggressive behavioural problems occurred. Moreover, it was no longer possible to make normal contact. Aged 3, at a specialised outpatient department for child and adolescent psychiatry, a diagnosis of autism spectrum disorder was made, at that time also called childhood disintegrative disorder. One year later, the patient was referred to a child neurology clinic for evaluation of his developmental regression. Somatic and neurological examination disclosed no abnormalities nor were there any dysmorphisms. Laboratory tests of relevant haematological and chemical parameters as well as extensive investigations, including MRI scanning of the brain, were all normal and a diagnosis of autistic retardation was made. In subsequent years, genetic, neurological, metabolic, ophthalmological and dermatological examinations were performed as well as a second MRI of the brain under general anaesthesia. Again, no aetiological explanation could be found for his developmental delay and the diagnosis of autistic retardation was confirmed. Because of his aggressive behaviour with temper tantrums, he stayed during daytime at an activity centre of an institute for people with intellectual disabilities where several short-lasting pharmacological interventions were tried that all had to be stopped because of increase of disinhibited and aggressive behaviours (promethazine, methylphenidate and phenobarbital) or motor side effects (risperidone). Apart from challenging behaviours, there were persistent sleep disturbances as well as intermittent urine incontinence and diarrhoea.
First neuropychiatric referral
At the age of 12, he was referred for specialised neuropsychiatric evaluation to the first author. Behavioural observation revealed clear ritualistic, stereotyped and autistic traits with mood instability necessitating structured individual guidance in all situations to avoid overestimation with subsequent challenging and aggressive behaviours. He could not read, write, calculate or tell the time. Somatic and neurological examination showed no abnormalities and there were no dysmorphisms. Weight, height and head circumference were in accordance with his biological age. Relevant haematological and biochemical parameters were all normal. The patient communicated with simple few word sentences, body language and pictorial signs. As assessed with the Vineland Adapative Behaviour Scale (VABS13) developmental age scores on the domains of communication, motor skills, socialisation and daily activities were 19 months, 42 months, 11 months and 24 months, respectively, corresponding with severe intellectual disability. At that time, he was treated by his mother with homeopathic compounds of which one also contained a very low dose of promethazine. For behavioural control, two times per day 0.4 mg haloperidol was added. To elucidate the aetiology of his severe intellectual disability, single nucleotide polymorphism (SNP) array analysis was performed that showed no abnormalities. Subsequently, specialised neuropsychiatric assistance was no longer needed.
Intermediate period
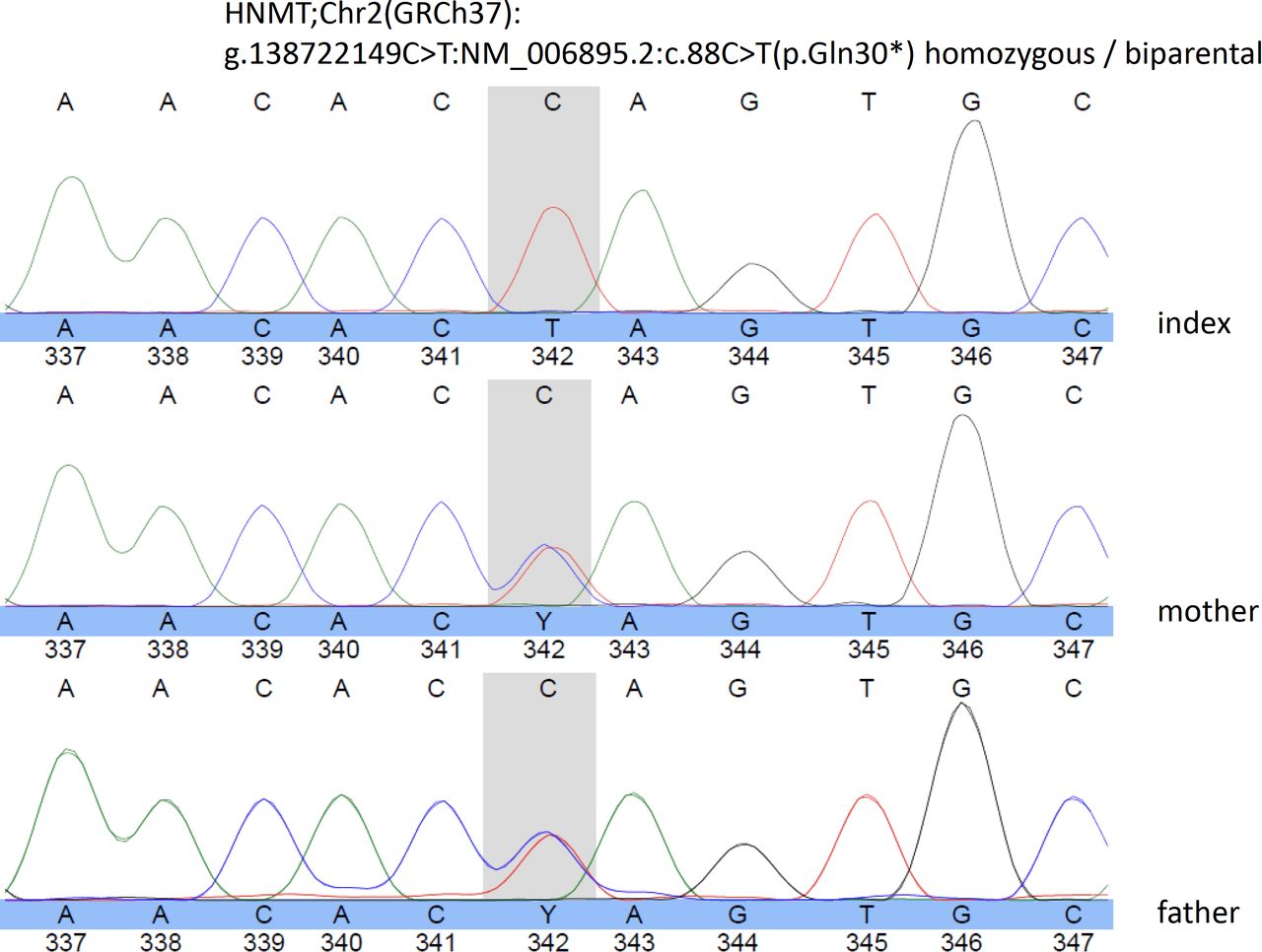
Aged 14, the patient developed severe headache, vomiting, neck flexion, urine incontinence and unstable gait on which haloperidol was discontinued. Since these symptoms worsened, multiple brain imaging was performed that demonstrated a brain tumour in the fossa posterior . He subsequently underwent fossa posterior craniotomy and a benign right-sided cerebellar haemangioblastoma was successfully removed (see figure 2 for preoperative MRI image of the brain and CT scanning 2 years later). Neurofibromatosis type I and Von Hippel-Lindau syndrome were genetically excluded. Thereafter, the patient functioned on his premorbid level without any neurological symptoms and returned to his parent’s home and continued during daytime the structured activity programme at his former institute for people with intellectual disabilities. Severe sleeping problems, intermittent urine incontinence and diarrhoea were still present. To improve his sleeping pattern, the treating physician prescribed 2 mg zuclopenthixol. Since no significant changes in his behaviour repertoire could be expected, neuropsychiatric consultation was not requested. However, at the age of 19, trio-based exome sequencing demonstrated a homozygous pathogenic nonsense variant in the first exon of the HNMT gene (g.138722149C>T:NM_006895.2:c.88C>T(p.Gln30*)) leading to a premature stop codon, which was confirmed by Sanger sequencing. Both parents were shown to be heterozygous carrier of this variant. Figure 3 shows the Sanger sequencing electropherograms of the index patient and his parents. These findings corresponded with a diagnosis of MRT51. Because of this finding, he was referred again for specialised neuropsychiatric evaluation.

(A) Aged 14 years. Preoperative T2-weighted-Fluid-Attenuated Inversion Recovery (FLAIR T2) MRI image of the brain showing right-sided cerebellar cystic tumour (cerebellar haemangioblastoma) with slight peritumoral oedema and a maximum cross-section of 56 mm, leading to compression of the fourth ventricle and slight displacement to the left (enhancing mural nodule and supratentorial obstructive hydrocephalus not visible). (B) Aged 16 years. CT scan image of the brain showing small residual cyst only with cross-section of 20 mm without any compression of the fourth ventricle.

Sanger sequencing electropherogram of the index patient, and his parents showing in the grey field the homozygous mutations of the index patient (C replaced by T).
Second neuropychiatric referral
At examination, aged 19, a similar behavioural repertoire was present as during the first consultation, aged 12, in that he still displayed autistic, ritualistic and challenging behaviours with aggressive acts and self injuries. Active language was restricted to single words or short and simple sentences. Somatic and neurological examination disclosed no abnormalities. Height (172 cm) and weight (70 kg) were in accordance with his current biological age. Relevant haematological (eg, white blood cell count and thrombocytes) and biochemical parameters (eg, vitamin status, thyroid and liver parameters, glucose and lipid spectrum) were all normal still. Pharmacological treatment comprised two times per day 40 mg pipamperone, irregularly combined with 15 mg mirtazepine or 7.5 mg midazolam to improve his sleep pattern. Also, two times per day 20 mg omeprazole was prescribed. Again, psychological assessment was performed. Social and emotional development as measured with the Dutch scale for emotional development in people with intellectual disability (ESSEON-R14) corresponded with a developmental age of 12 months and 18 months, respectively. As assessed with the VABS, developmental age scores on the domains of communication, daily activities and socialisation were 23 months, 23 months and 15 months, respectively, being not significantly different from those as established previously. With the Dutch scale for social life skills (SRZ15), a developmental age of 2–3 years was established, corresponding with a cognitive level of 2–4 years, enhancing the risk of overestimation. He was then referred to a university outpatient department for child and adolescent psychiatry where the diagnosis of autism was confirmed. Subsequently, he attended the outpatient department of clinical pharmacology for treatment advice.
Treatment
Because of the demonstrated homozygous mutation in the HNMT gene resulting in the complete absence of a functional HNMT, treatment with the antihistaminergic drug hydroxyzine in a daily dose of 25 mg and a histamine-restricted diet was prescribed. Since their efficacy was doubtful, it was advised to stop the use of psychotropics as mentioned before with the exception of omeprazole.
Outcome and follow-up
Until now, aged 23, treatment with 25 mg hydroxyzine in combination with a histamine-restricted diet resulted in normalisation of the patient’s sleep–wake cycle, significant reduction of aggression, improvement of speech and receptive language capacities, and complete continence for urine and faeces. The patient (figure 4) still lives at his parents’ home following, like in previous years, during daytime an activity programme at the same institute for people with intellectual disabilities.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Picture of the patient without any dysmorphic features: (A and B) aged 17 years and (C and D) aged 23 years.
Discussion
Here, a 23-year-old severely intellectually disabled Dutch male patient born from second cousins is described in whom trio-based exome sequencing demonstrated a homozygous mutation in the HNMT gene matching a diagnosis of MRT51. Apart from one male patient with a homozygous mutation of the HNMT gene but without any phenotypic description (DECIPHER (324 002)), to the best of our knowledge, this is the first patient with this genetic syndrome after the publication of seven individuals from two unrelated families of Turkish and Kurdish descent, respectively.12 Like in the affected members of these two families, also in our patient, global regression occurred around the age of 4 years, most pronounced regarding speech and language, in the absence of any dysmorphic features or congenital anomalies. Although not explicitly mentioned in the description of the patient histories of the two unrelated families from Turkish and Kurdish descent, it can be assumed that in all, like in our patient, a diagnosis of autism could have been made.
Unfortunately, in the publication of Heidari and coworkers, no information is given about either sleep pattern and intestinal problems or advised treatment regimen. In the here described patient, treatment with the antihistaminergic compound hydroxyzine in combination with a histamine-restricted diet resulted in normalisation of sleep pattern, complete continence, improvement of active speech and a significant reduction of aggressive challenging behaviours.
In conclusion, the behavioural phenotype of HNMT-associated MRT51 may comprise not only regression with loss of earlier achieved capacities around the middle of the first decade, but also autism, dysregulation of sleep–wake cycle and intestinal problems. The latter two may be effectively treated with the antihistaminergic compound hydroxyzine in combination with a histamine-restricted diet. Because of the attained marked and long-lasting improvement of this patients’ general functioning, in retrospect, the earlier postulated regression hypothesis may have to be reconsidered.
Patient’s perspective
We, the parents of the described patient, have seen a significant improvement in general functioning after the diagnosis was established and our son started the medication in combination with the histamine-restricted diet. He is now much calmer, sleeps well, shows no more aggression and has become completely potty-trained. It is also noticeable that he has pleasure again in his activities within the daily activity centre. Finally, we as well as his institutional supervisors can make with simple terms much better contact with him. We are very happy with all these positive developments.
Learning points
Medical professionals should consider whole exome sequencing as the starting point for aetiological investigation.
Brain histamine is crucial in physiological functions like sleep and stress regulation.
Histamine N-methyltransferase deficiency is associated with aggressive behaviours and mental retardation, autosomal recessive 51.
Antihistaminergic compounds that pass the blood–brain barrier such as hydroxyzine, combined with a histamine-restricted diet, normalise sleep pattern, reduce levels of challenging behaviour and ameliorate communication.
Acknowledgments
Written informed consent was obtained from the parents for publication of the case history of the patient; the parents also kindly provided the picture of the patient at present age. The patient was referred by the Centre for Consultation and Expertise, region West. The authors are indebted to the staff members of the Gemiva-SVG Institute for people with intellectual disabilities, location Zoeterwoude, for their careful observations of the behavioural status of the patient and the psychological assessments. For subsequent advises about treatment, the patient was referred to the outpatient department of clinical pharmacology at the Erasmus Medical Centre Rotterdam. MRI and CT images were reevaluated and selected by Dr GA Hoffland, (neuro)radiologist at the VieCuri Medical Centre, Venlo, the Netherlands.
Footnotes
Contributors WMAV and JIME conceptualised and designed the study and reviewed the literature. WMAV assessed the patient, acquired the data and discussed the initial findings. WMAV and JIME reported the case history and drafted the manuscript. PKCJ commented on the literature review, initially evaluated the manuscript and created the figure showing the histamine neurotransmission process. AvH interpreted the genetic data and provided the Sanger electropherogram as well as the picture of the patient aged 17 (A and B). PKCJ and AvH critically reviewed the manuscript. All authors read and approved the final version of the manuscript.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Patient consent for publication Parental/guardian consent obtained.
Provenance and peer review Not commissioned; externally peer reviewed.