Article Text

Abstract
Yolk sac tumour (YST), the most common germ cell tumour (GCT) in infants and children, accounts for 80% of GCTs in this age group.1 It is observed in only 2.4% of adult patients in their pure form since up to 42% of mixed GCTs have some component of the yolk sac.2 We present a 46-year-old cryptorchid man who presented with complaints of abdominal lump, pain and altered bowel habits since 1 month. CT scan of the abdomen revealed a mass arising from the pelvic cavity with feeders predominantly from the left gonadal vessel. Mass was excised via exploratory laparotomy and sent for histological examination. Microscopic examination was suggestive of pure YST. Immunohistochemistry stained positive for cytokeratin, placental-like alkaline phosphatase, Glypican 3 and alpha fetoprotein. The patient succumbed prior to commencing chemotherapy. Pure YSTs of post-pubertal origins are extremely rare and are more aggressive than their prepubertal counterparts.
- surgical oncology
- oncology
- pathology
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Background
Testicular cancer accounts for only 1% of all male cancers.3 Germ cells being the most abundant in the testes, it is not surprising that germ cell tumours (GCTs) are the most common type of testicular cancer overall accounting for 95% incidence. Of all the GCTs, 50% comprise seminomas and 50% are nonseminomatous GCTs.
Pure yolk sac tumour (YST) is primarily found in infant and childhood age group and accounts for 80% of testicular tumour in this age group; however, it is a very rare entity in pure form in adulthood accounting for just 2.4% of adult age group patients presenting with testicular lesion.4
We present a case of pure YST, post-pubertal type, occurring in a man with bilateral intra-abdominal cryptorchid testes. Few cases regarding pure YST, post-pubertal type, of testicular origin have been reported in literature, but none in bilateral intra-abdominal cryptorchid testes. Thus, we believe that this case is the first of its kind.
Case presentation
A 46-year-old married man was referred to the oncosurgery department in April 2018 with complaints of gradually progressive, mildly tender, abdominal lump of insidious onset, since the last 1 month. Other significant contributory history included the fact that the patient had delayed bowel movements since 15–20 days, as well as one to two episodes of non-projectile non bilious vomiting—1 week prior to admission—with the fact that the patient has bilateral cryptorchidism and was infertile and not on any treatment for the same. On detailed interrogation, the patient gave history of progressive painless diminution of bilateral visual fields over the course of 10 years.
On examination, the patient presented with a single, ovoid, non-tender, firm, non-ballottable, abdominal lump approximately 25 cm × 15 cm, predominantly occupying infra-umbilical segment of the abdomen, with pelvic extension. Percussion revealed a tympanic note in the supra-umbilical region and a dull note in infra-umbilical region. Auscultation was unremarkable. Per rectal examination was normal. Examination of the scrotum revealed absence of both testes within the scrotal cavity.
Additionally, the patient did not present with features of feminisation.
Ophthalmological examination revealed that visual acuity had reduced to hand movements with typical findings of optic atrophy on fundoscopic examination.
Investigations
Apart from neutrophilic leukocytosis on haemogram and presence of calcium oxalate crystals on urine examination, all other routine blood investigations were within normal limits.
Serum α-fetoprotein level was >15 160 ng/mL, which was significantly elevated. Serum β-HCG level was 2 mIU/L and serum LDH was 198 U/L, both of which were within normal limits.
Contrast-enhanced CT scan of the abdomen was suggestive of a large, well-defined, heterogeneous soft tissue density mass in the hypogastric supra vesical region measuring about 7.8 × 9.2 × 11.2 cm. Post contrast study shows moderate heterogeneous enhancement with significant irregular non-enhancing necrotic areas within. Predominant tumorous feeders are seen arising from the left gonadal artery. The lesion displaces the small bowel loops superiorly and extends laterally to the right iliac fossa, anteriorly to the right common iliac and external iliac vessels. Superiorly, there is a small exophytic tumorous extension seen into the mesenteric sleeve. Inferiorly, the lesion indents the dome of the bladder with thin intervening fat planes. There is a well-defined hypodense nodular structure seen in the right iliac fossa measuring about 2.5×3.0 × 2.7 cm inferior to the base of caecum. Post-contrast study shows minimal heterogeneous enhancement with arterial supply arising from right gonadal artery. Both scrotal sacs are empty suggestive of undescended testes (figure 1).

Transverse, coronal and sagittal sections of contrast-enhanced CT of abdomen and pelvis.
Non-contrast brain CT revealed normal study.
Ultrasound-guided tru-cut biopsy was positive for malignant cells and favoured seminoma. Chest radiograph and 2D echocardiogram were normal.
Differential diagnosis
Seminoma
Treatment
On the fifth post-admission day the patient underwent prophylactic bilateral DJ stenting, and on the eighth post-admission day the patient underwent exploratory laparotomy under general anaesthesia after giving informed consent. Intra-operative findings were consistent with large irregular greyish-white to grey yellow to grey brown mass seen arising from pelvic cavity (figure 2). The mass was extremely friable to touch, with variable tissue consistency, due to which it could not be removed completely. The mass was fixed to bladder dome and base, as well as to two to three segments of jejunum and ileum, without evidence of perforation. The second mass (right cryptorchid testis) could not be separately identified from the mass. Around 1700 g gross weight of tumour was removed, leaving behind fixed areas as mentioned above. Haemostasis was achieved, after which closure commenced. Post-operative recovery was uneventful apart from the fact that the patient was transfused with 2 pints of packed cell volume.

Gross specimen of tumour.
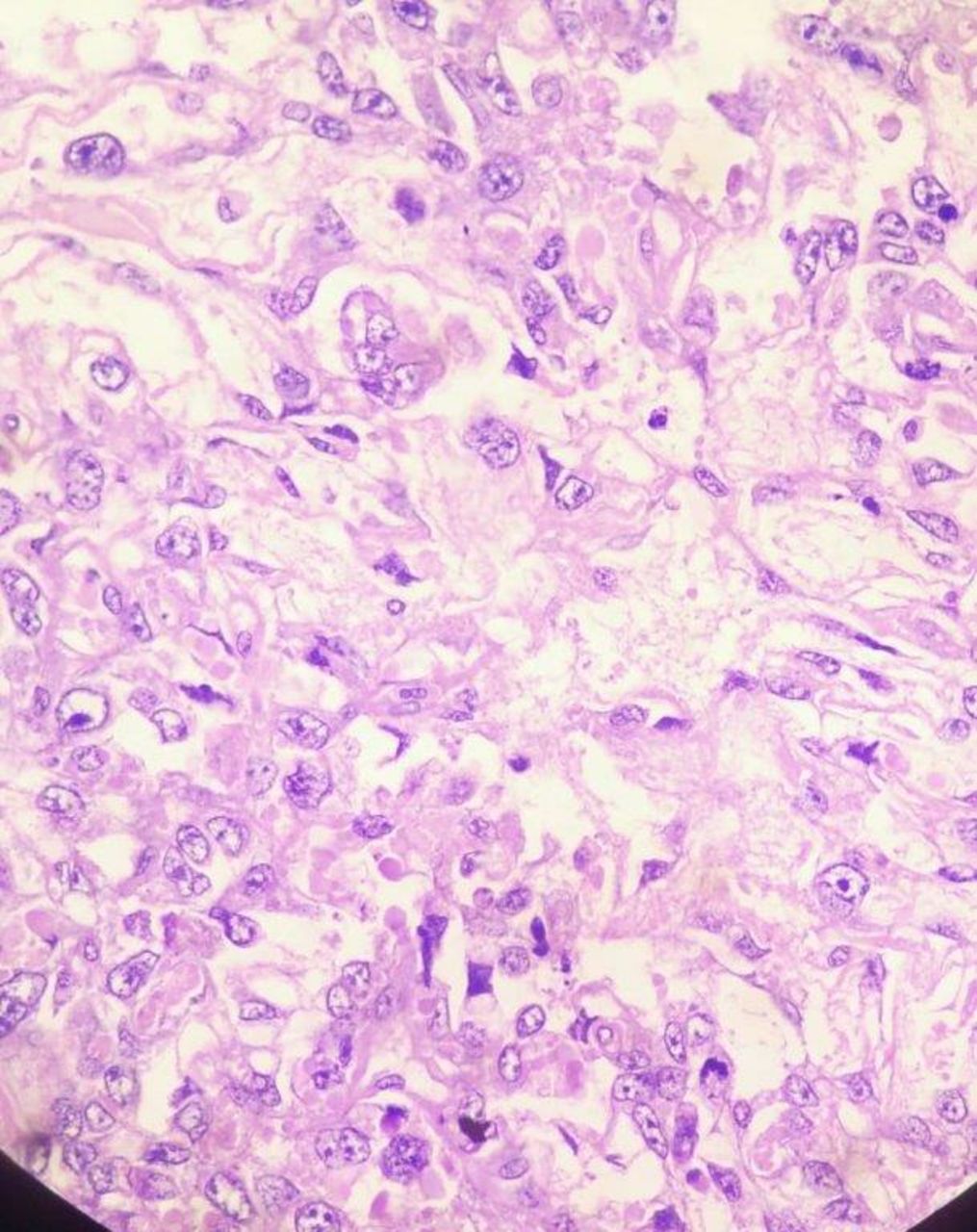
Histopathology examination of cut section showed tumour cells in an alveolar to endodermal sinus to microcystic pattern. Most of the tumour cells had large pleomorphic vesicular nuclei with predominant nucleoli and meagre cytoplasm. Microcystic areas were composed of flattened cells.
Schiller–Duval bodies, hyaline globules, areas of necrosis and haemorrhage were noted (figures 3–5).

Schiller–Duval body.
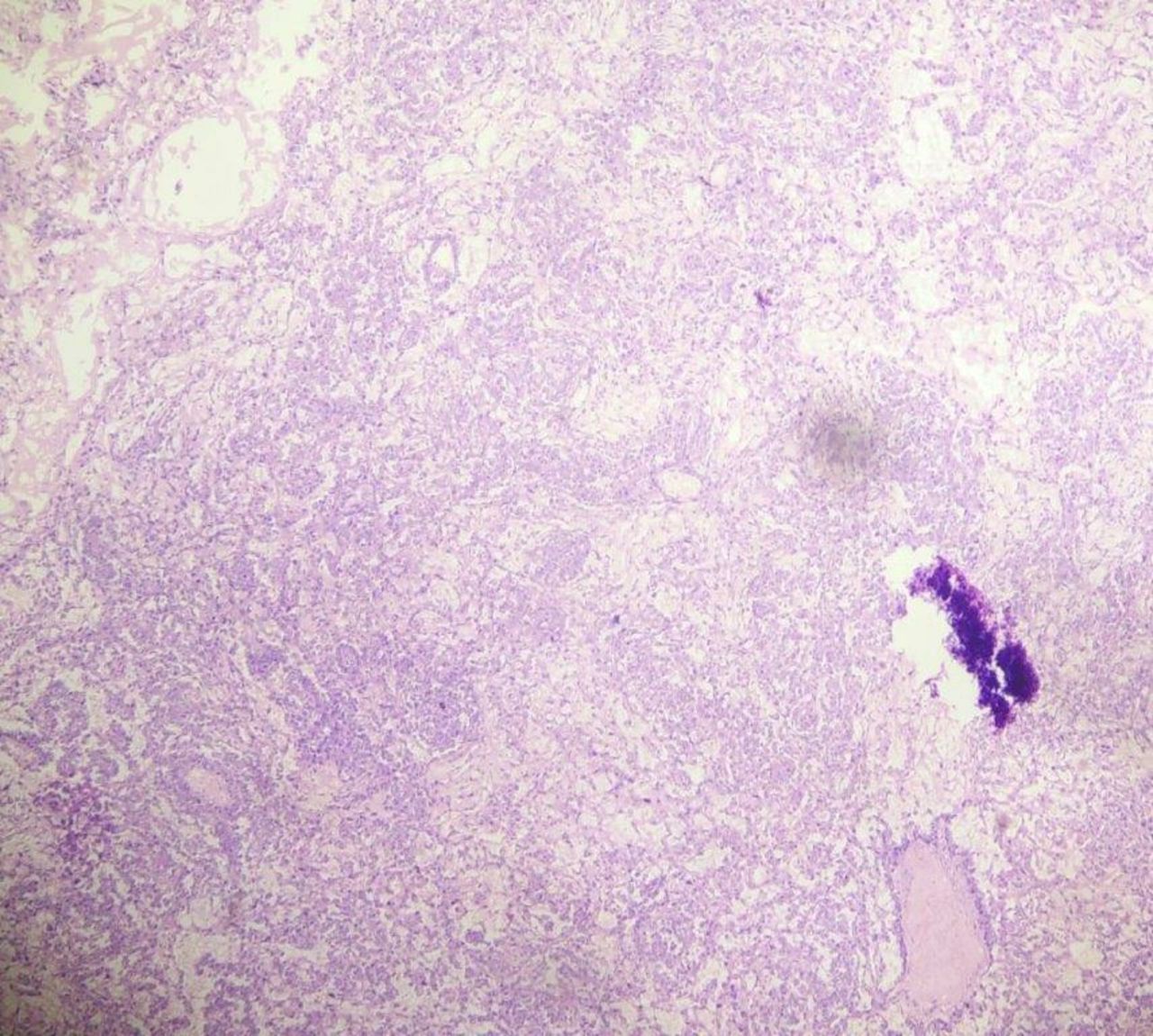
H&E stain under 10x magnification.
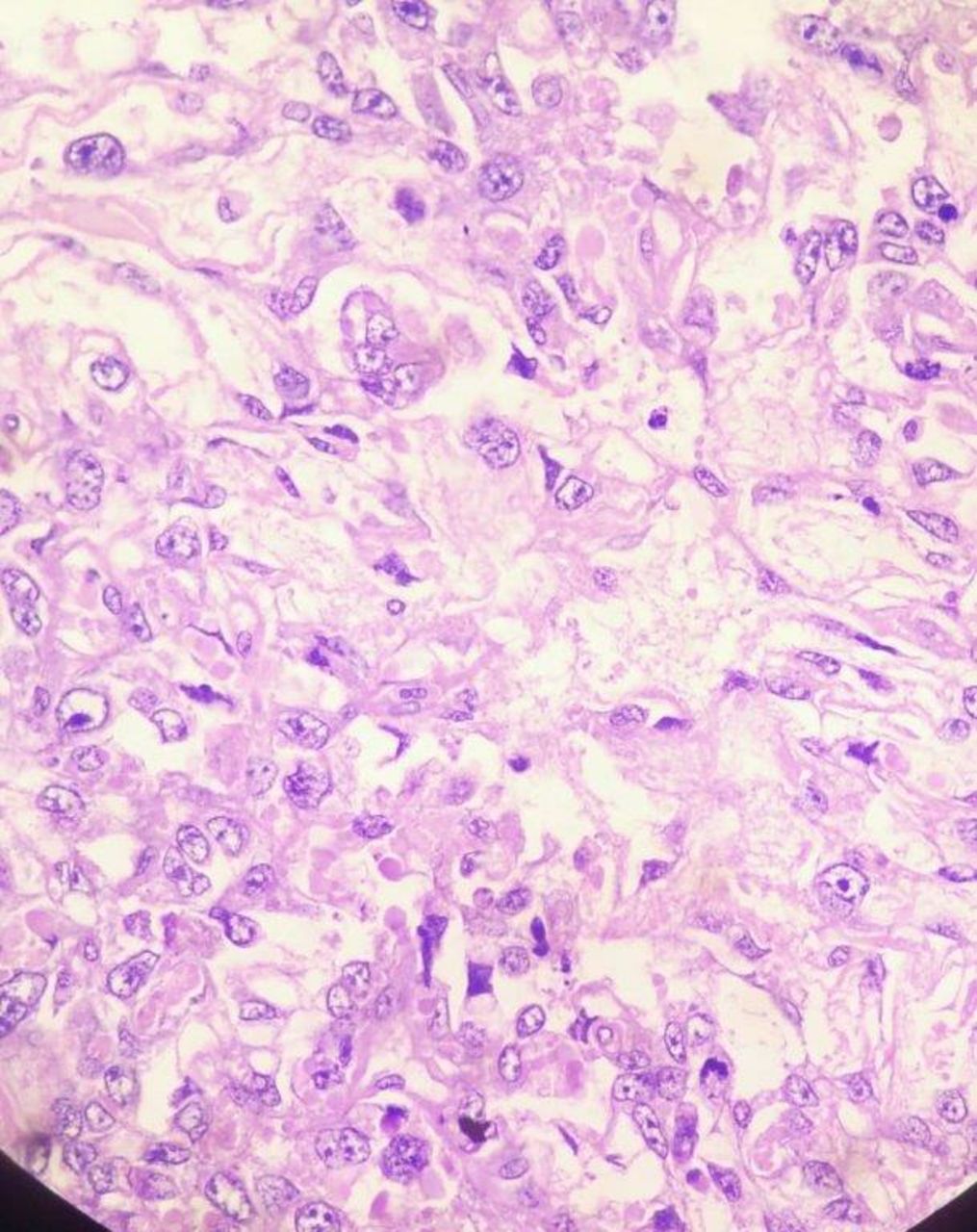
H&E stain under 40x magnification.
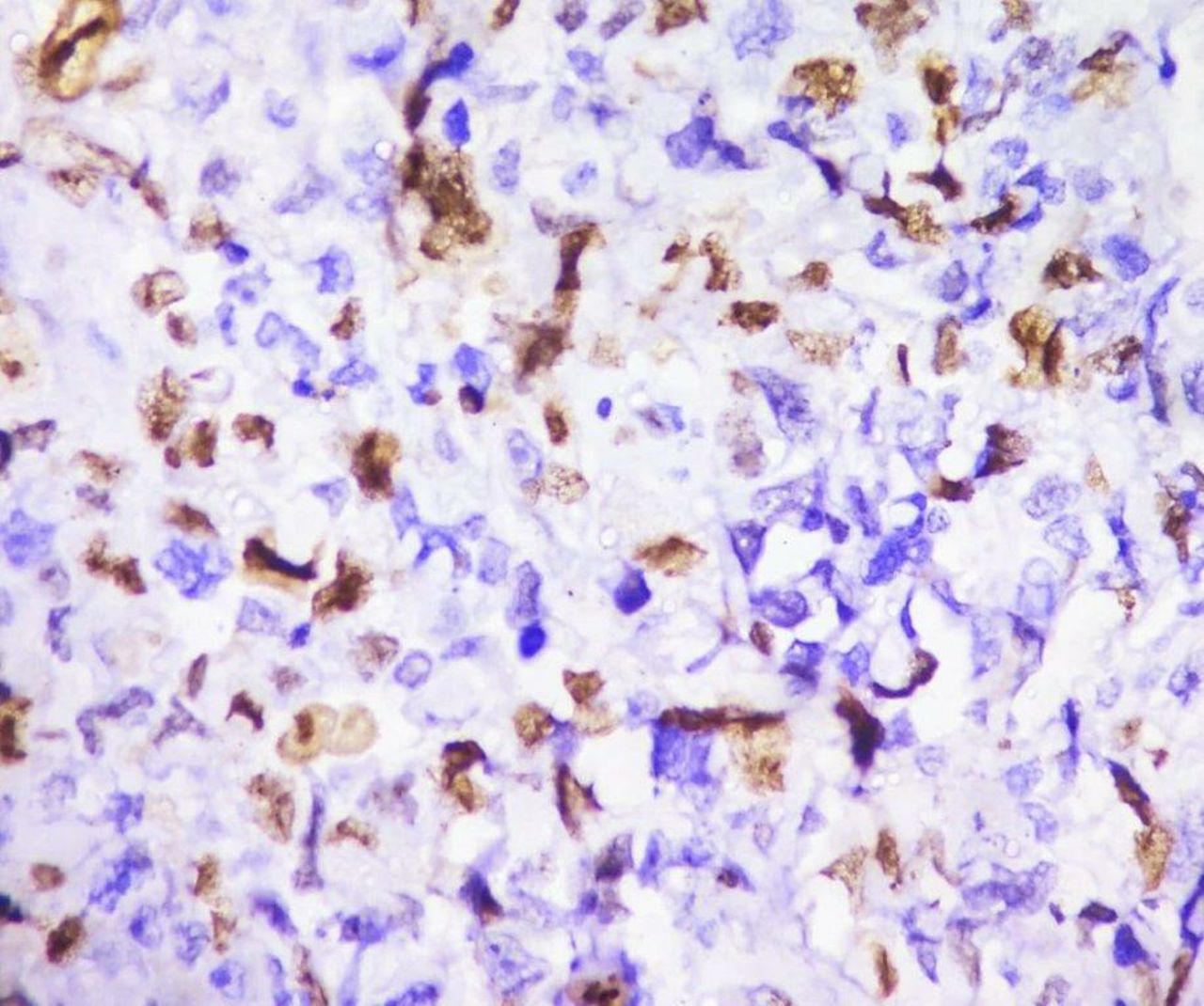
Above features were characteristic for pure YST. Immunohistochemistry report revealed diffuse strong positive for cytokeratin (CK), patchy strong positive for placental-like alkaline phosphatase, moderate positive for Glypican 3, moderate patchy positive for alpha fetoprotein (AFP), ki-67 proliferative index was 30%, negative for epithelial membrane antigen (EMA), Oct3/4 and CD30 (table 1) (figures 6–10).

Immunohistochemistry for cytokeratin—diffuse strong positive in tumour cells.

Immunohistochemistry for placental-like alkaline phosphatase—patchy strong positive in tumour cells.
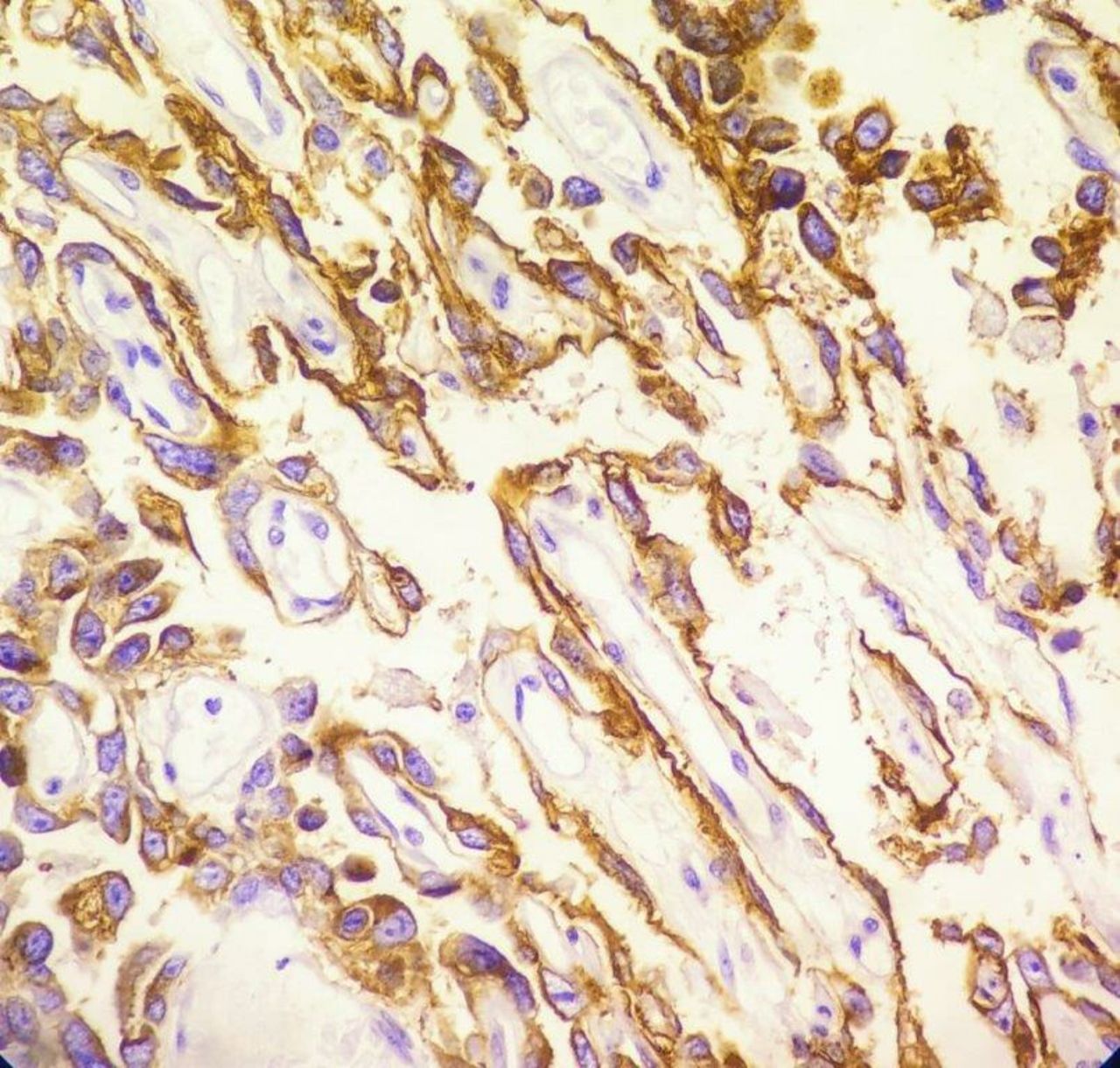
Immunohistochemistry for Glypican 3—moderate positive in most tumour cells.

Immunohistochemistry for alpha fetoprotein—moderate patchy positive in tumour cells.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Immunohistochemistry for Ki-67—proliferative index is 30%.
Immunohistochemistry report of the tumour
Impression: Yolk sac tumour, post-pubertal type.
The patient was discharged on seventh post-operative day. After surgical de-bulking, the plan was to initiate chemotherapy with curative intention.
Outcome and follow-up
However, 2 weeks after discharge, the patient was readmitted with abdominal pain and discharge through suture line which was serosanguinous and non-foul smelling. The decision was made not to re-explore the patient as he did not present with signs and symptoms of bowel obstruction. Over the course of the next week, the patient turned severely cachexic, discharge increased and patient developed septicaemia, which was refractory to treatment. Ten days after re-hospitalisation, the patient succumbed to septicaemic shock, without even initiating the first chemotherapy cycle.
Discussion
YSTs are a class of GCTs that resemble yolk sac, allantois and extra-embryonic mesenchyme. We are still in the dark about the origins of pure YST in adults as against hypermethylation of RUNX3 promoter gene and overexpression of GATA-4 genes which are active in prepubescent type. 12pi, aneuploidy and p53 mutations are noted in adults as opposed to other genetic mutations and chromosomal aberrations found in prepubescent type. Metastasis occurs only through lymphatic spread in adults as opposed to lymphatic and haematogenous spread in prepubescent type.5
Statistics state that incidence of testicular cancer is threefold to fivefold in population with cryptorchidism and that incidence decreases if correction orchidopexy was done below the arbitrary age of 13 years.6 7 It is noteworthy in mentioning that in cases of unilateral cryptorchid testis, the undescended and the normally descended testis are at increased risk of developing malignancy.8 Incidence of seminoma in cryptorchid testis far outweighs that of YST.
YSTs of testicular origin usually present as large painless bulky masses. Almost all the patients with a YST, either in pure form or mixed form, have a significantly elevated serum alpha fetoprotein.
Grossly, YSTs are solid grey-white with a gelatinous, myxoid or mucoid appearance. Necrosis, cystic changes and haemorrhage are often seen. In adults, the gelatinous appearance of these tumours is mingled with the gross findings of the other GCT components; therefore, the overall appearance of a YST is heterogeneous.
Microscopically, the appearance of YST is the same regardless of patient age and primary site. Several different patterns are usually present in varying proportions, though one pattern may predominate. YSTs that present with only one histological pattern are exceedingly rare. Some of these patterns include microcystic/reticular pattern, macrocystic pattern, solid pattern, glandular-alveolar pattern, endodermal sinus pattern, papillary pattern, myxomatous pattern, polyvesicular vitelline pattern, hepatoid pattern and enteric pattern. Schiller–Duval bodies are a hallmark of YSTs seen in 50%–75% cases (figure 3). YSTs contain intracellular and extracellular PAS-positive hyaline globules.9 10
Immunohistochemically, YSTs are positive for AFP, CK AE1/AE3, Glypican-3 and SALL4 and are negative for EMA and CK7. A negative CD30 and beta-HCG on immunohistochemistry can help rule out the mixed components of embryonal carcinoma and choriocarcinoma, respectively.11
Nonseminomatous tumours in the advanced stages are considered to be poor prognosis when associated with any of the following criteria: visceral metastases, alpha fetoprotein >10 000 ng/mL, β-HCG >50.000 IU/L or LDH >10× upper limit of normal.12
As per the prognostic classification of metastatic seminoma and nonseminoma according to International Germ Cell Cancer Collaborative Group, our case has gonadal primary tumour location with elevated levels of serum markers, showed no extrapulmonary/visceral metastasis and hence belonged to the group that has poor prognosis (48% survival rate).
The primary treatment of choice for advanced disease is three to four cycles of PEB combination chemotherapy (cisplatin, etoposide, bleomycin), followed by resection of residual tumour.
As per above discussion, in our case, much of the gross, microscopic and immunohistochemistry examinations were consistent with that of YST. Surgical debulking in our case was carried out prior to chemotherapy, with the intent of relieving patient’s symptoms.
Patient’s perspective
Patient’s relatives: we are extremely sad and disheartened at the unfortunate demise of our beloved brother. We hope that his rare ailment might spark new research activities and better/newer treatment regimes to a possible cure for this aggressive disease.
Learning points
Pure yolk sac tumour is a rare entity in adult patients, and when diagnosed, should undergo close follow-up as it is associated with poor prognosis in said age group as against its prepubescent counterpart.
Importance of testicular examination in every abdominal case cannot be stressed enough.
In closing, the diagnosis of pure yolk sac tumour, post-pubertal type, should be made only after meticulous microscopic examination rules out other germ cell components.
Acknowledgments
The authors sincere thank to pathology department of Krishna Institute of Medical Sciences and Dr Rajni Parmar and Dr Aniruddha Chattoraj for contributing excellent quality pictures.
Footnotes
Contributors HJ urged us to publish this article in view of the rare nature of this tumour. Under his expert guidance, the tumour was excised. He has added to the literature of the article. HJ, JM and SG have contributed to acquiring data and management of the patient as well as gathering information and in-depth research into yolk sac tumours.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Patient consent for publication Next of kin consent obtained.