Article Text

Statistics from Altmetric.com
Description
Pachydermoperiostosis (Touraine-Solente-Golé Syndrome) is a hereditary disorder which usually presents with bony lesions (periostosis), digital clubbing and skin thickening (pachyderma). Facial coarsening, spade-like hands and feet with increased sweating may cause diagnostic confusion between acromegaly and pachydermoperiostosis. We report a case of an 18-year-old man who presented with progressive enlargement of hands and feet, increased sweating of palms and sole and drooping of eyelids since birth. Family history of similar illness was present in his maternal aunt and grandfather. Pachydermoperiostosis was suspected and evaluated further by hormonal workup and radiological studies. We made a diagnosis of complete (classic) pachydermoperiostosis based on the findings of normal insulin- like growth factor-1, hyperhydrosis, blepharoptosis, skin thickening, digital clubbing and prominent radiological abnormalities.
An 18-year-old man, resident of Jharkhand, India, presented with chief complaints of progressive disproportionate enlargement and excessive sweating of hands and feet since birth (figure 1A,B). He was born out of a non-consanguinous marriage at term with a birth weight of 2.6 kg. The antenatal and perinatal periods were uneventful. His developmental milestones and scholastic performances were normal. He did not give any history of palpitations, heat intolerance, weight loss or hyperdefecation. Neither were there any history of headache, visual disturbances or other features of anterior pituitary hormone insufficiency. However, there was family history of similar enlargement hands and feet in his maternal aunt and maternal grandfather.

(A and B) Showing excessive soft tissue in hand and feet with clubbing.
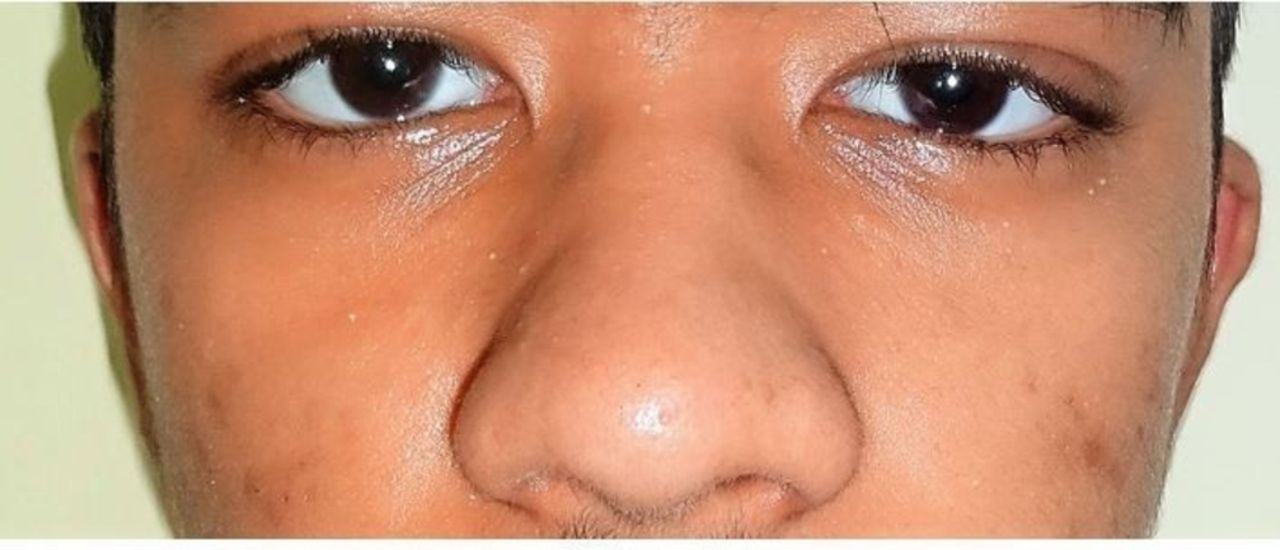
On examination, he was normotensive with a height of 163 cm (height SD −2.1, mid parental height of 166.5 cm), weight of 54 kgs (5th–10th centile) and upper segment:lower segment ratio of 0.87. He had normal secondary sexual characteristics with a Tanner’s staging of G4, P4 and A+. His hands and feet were enlarged with bilateral knee swellings. He had hyperhidrosis of both palms and soles with bilateral fine tremor. He had no prognathism but had malalingment of teeth. There was no goitre or gynaecomastia. He also had slight drooping of both eyelids and a bulbous nose (figure 2). Grade 4 clubbing was present in both hands and feet (figure 1A, B).

Showing slight drooping of both eyelid and bulbous nose.
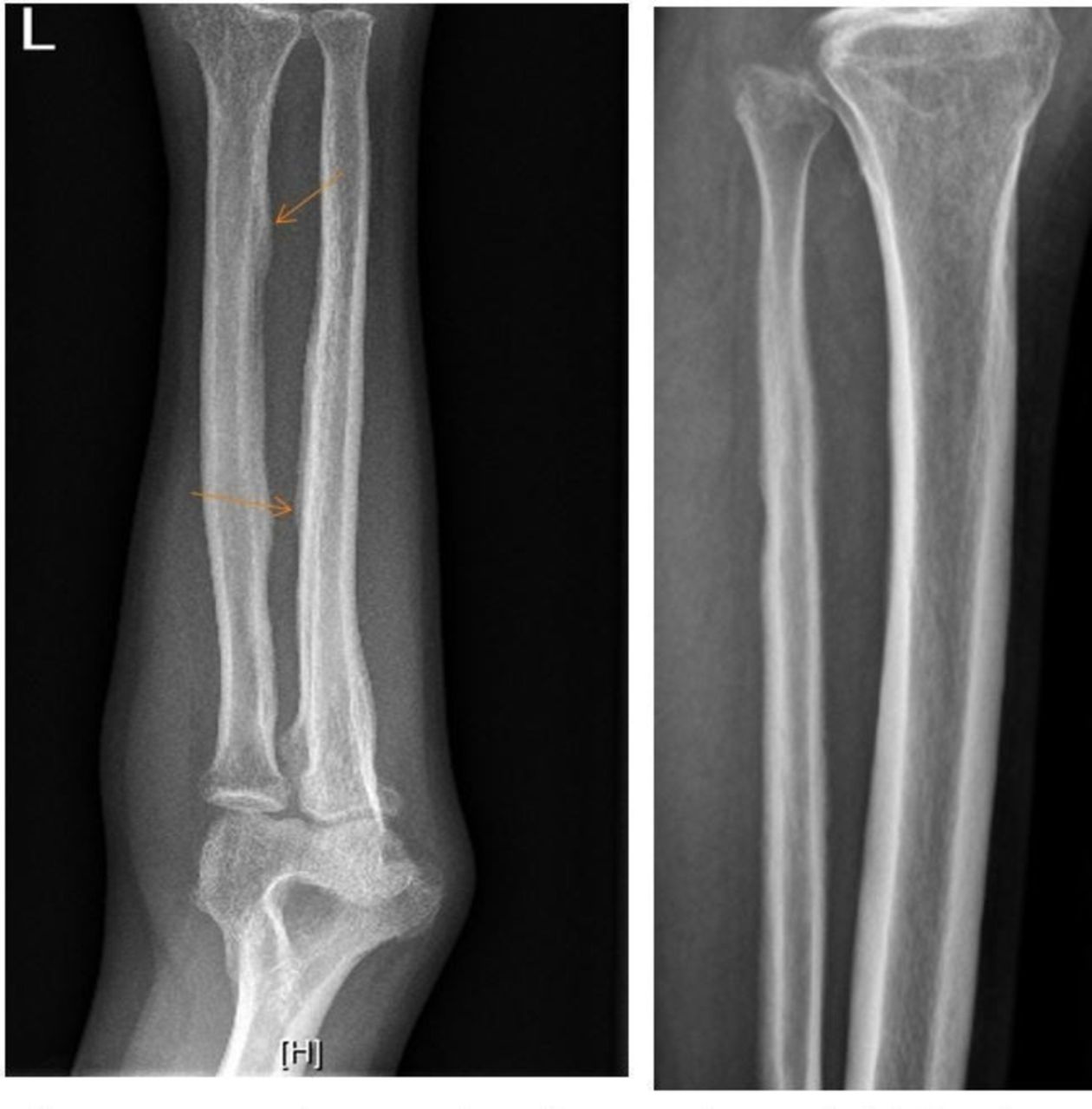
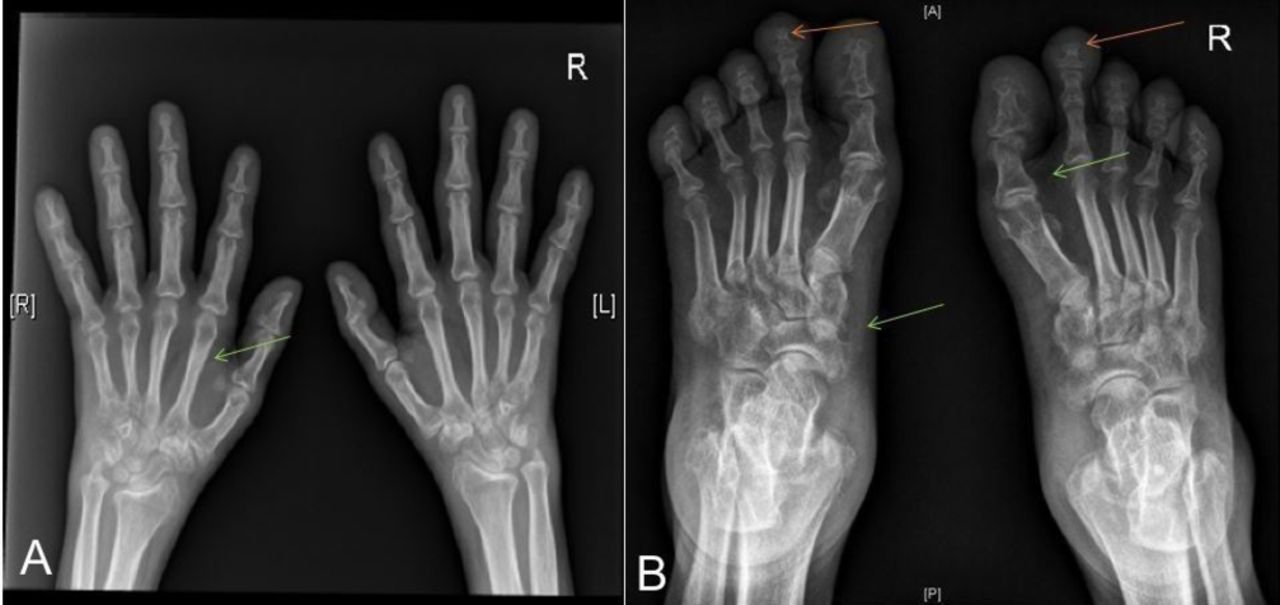
Systemic examination was essentially normal. Further investigations revealed the presence of mild iron deficiency anaemia without any other peripheral blood count abnormalities. He had normal liver function tests, serum creatinine and electrolytes; hormonal evaluation revealed thyroid stimulating hormone levels of 3.2 mIU/L (N=0.3–4.5) with total T4=10.2μg/dL (N=4–12) and free T4=1.19 ng/dL (N=0.8–1.8). The Insulin-like Growth Factor(IGF)-1–210 ng/mL (normal range, 109–527) levels were normal with suppression of growth hormone after 75 g oral glucose to 0.12 ng/mL (normal <0.4). Serological workup for connective tissue diseases and vasculitis was negative, while there was presence of vitamin D deficiency (25-OH vitamin D=9.6 ng/mL, normal >30). Radiological evaluation of long bones was suggestive of periostitis mainly in the diaphyseal region (figure 3). X-ray of both hands and feet were suggestive of extensive soft tissue enlargement and evidence of periostitis and acro-osteolysis (figure 4A, B). While a normal upper gastrointestinal (GI) endoscopy ruled out the possibility of gastric ulcerations or erosions leading to blood loss, cardiopulmonary causes for the condition were excluded based on normal investigations including an ECG, echocardiogram, arterial blood gas analysis, chest X-ray and pulmonary function tests. Based on these clinical, biochemical and radiological findings, a diagnosis of classic (complete) pachydermoperiostosis with mild iron deficiency anaemia of nutritional aetiology was made.

Long bones showing periosteal thickening mainly in the diaphyseal region (arrows).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
(A and B) Showing excessive soft tissue in hand and feet with evidence of periostitis (green arrow) and acro-osteolysis (first and second toe, orange arrow).
Pachydermoperiostosis was first described by Friedreich in 1868,1 who called it ‘Hyperostosis of the entire skeleton’. The estimated prevalence of the disease is 0.16%2 with symptoms usually appearing around puberty, with a male to female ratio of 7:13 and severely affects men. Although an autosomal dominant model with incomplete penetrance and variable expression being proved,4 both autosomal recessive and X-linked inheritance patterns have been suggested. An infantile form has also been described5 which is characterised by early presentation with enlargement and delayed closure of the cranial sutures, patent arterial duct and skin manifestations. This condition progresses slowly for a few years and is self-limiting thereafter.
This disease may present in three forms: classic or complete (skin thickening, skeletal changes and digital clubbing), incomplete (skeletal changes without involvement of skin) and forme fruste (skin thickening with minimal skeletal change). The major criteria includes periostosis, pachyderma and digital clubbing.6 The minor criteria mentioned in the literature are blepharoptosis, hyperhidrosis, arthralgia, seborrhea, joint effusion, gastric ulcer, cutis verticis gyrate, flushing oedema, acne, column like legs. Our patient had three major criteria (digital clubbing, bony changes on skeletal radiograph and skin thickening) and three minor criteria (joint effusion, sweating and blepharoptosis) which was sufficient to label him as a case of complete(classic) pachydermoperiostosis. The exact cause is unknown but recently mutations in 15-OH prostaglandin dehydrogenase on chromosome 4q34.1 has been suggested as the possible aetiology.7 Genetic analysis was planned in our case, but could not be performed due to financial constraints of the patient.
As the patient did not complain of any visual symptoms or cosmetic concerns regarding drooping of the eyelids, active intervention was deferred and patient was advised to follow-up periodically. Differential diagnosis for this rare condition includes syphilitic periostitis, thyroid acropachy, acromegaly, Van Buchem’s disease, diaphyseal dysplasia, secondary hypertrophic osteoarthropathy and chronic inflammatory rheumatic diseases.
A range of benign and malignant disorders8 have been reported in association with pachydermoperiostosis. These include facial epidermoid carcinoma, hypertrophic gastritis, peptic ulcer, gastric adenocarcinoma, Crohn’s disease and myelofibrosis. As a consequence of increased soft tissue bulk and hyperostosis, complications may arise such as ptosis, compression of the nerve endings, hearing problems, kyphosis, arthrosis, osteonecrosis of the femoral head and carpal tunnel syndrome. Our patient did not have any of these complications. The anaemia seen in these patients can be multifactorial—including myelofibrosis, gastric ulcers and GI bleeding and a possible serum inhibitor of erythropoetin.9 None of these were present on investigations in our patient and the iron deficiency anaemia was attributed to nutritional deficiency. He was supplemented with oral iron therapy and planned for follow-up.
No specific treatment is currently available. Medical management10 is indicated for symptomatic benefit and it includes non-steroidal anti-inflammatory drugs, colchicine, corticosteroids, retinoids, tricyclic antidepressants, risedronate, pamidronate and tamoxifen citrate to control arthritis. Botulinum toxin-A has also been tried for cosmetic reasons. Surgical management may include correction of bony deformities if any, and plastic surgery for disfigurement. Prognosis for this disease is good and patients have normal life expectancy. Our patient was advised to be on regular follow-up and is currently only on oral iron and vitamin D supplements in view of anaemia and vitamin D deficiency.
Learning points
The combination of digital clubbing, pachyderma and periosteitis should warrant consideration of pachydermoperiostosis after a thorough evaluation to rule out cardiopulmonary, hepatic and mediastinal disorders.
A combination of major and minor clinical features makes the diagnosis more likely, though differentials including thyroid acropachy, acromegaly and rarer causes like Van Buchem’s disease need to be carefully considered.
Family medical history, specially if autosomal dominant, should prompt to think of pachydermoperiostosis rather than acromegaly, though acromegaly can less commonly be familial.
Footnotes
Contributors All the authors have contributed to the manuscript and declared no conflict of interests. SA and RD have contributed to the data collection, diagnostic workup, review of literature, preliminary writeup and finalising the manuscript for submission. TVP and NT have contributed to the diagnostic workup, review of literature and finalising the manuscript for submission.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Patient consent for publication Obtained.