Article Text

Abstract
Haemophilia is a hereditary X-linked recessive disorder caused by a deficiency of either clotting factor VIII (haemophilia A) or IX (haemophilia B). Conventional treatment is currently based on the use of either plasma derived or recombinant coagulation factors. This paper reports on the case of a patient with severe haemophilia who presented with mesial decay and interproximal tartar build-up, for which extraction and scaling to remove tartar deposits were indicated. Following extraction, the usual haemostasis techniques were applied, and postoperative prophylactic antihaemophilic treatment was indicated for 2 or 3 days. The patient presented with moderate bleeding for a few minutes immediately after the procedure. Administration of factor VIII before surgery as well as the patient’s favourable pharmacokinetic response allowed for an optimal result. This treatment has afforded patients with haemophilia a better quality of life, and safe and efficient access to invasive surgical procedures.
- haematology (incl blood transfusion)
- dentistry and oral medicine
- pharmacology and therapeutics
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Case presentation
Haemophilia is a hereditary X-linked recessive disorder caused by a deficiency of either clotting factor VIII (haemophilia A) or IX (haemophilia B).1 2 The prevalence of haemophilia A (also known as classic haemophilia) is estimated at 1 in 6000 men while that of haemophilia B (or Christmas disease) has a prevalence of 1 in 30 000 men. The size of cDNA is 7056 base pairs (bp) for factor VIII and 1389 bp for factor IX. Consequently, the size of the protein is 280 kDa for factor VIII and 68 kDa for factor IX. The reference plasma level is 200 ng/mL for factor VIII and 5000 ng/mL for factor IX. The half-life of these proteins is very short, which means they have a high turnover (around 12 hours for factor VIII and 20 hours for factor IX). These proteins are mainly synthesised in the liver and, to a lesser extent, in the kidney and endothelium. The pathological condition is considered severe when factor plasma levels fall below 1% of normal; moderate when levels range between 1% and 5%; and mild when factor activity is between 5% and 40%. The overall prevalence of the disease is 7.7/100 000, which means it is considered a rare haematologic disorder (Orpha No ORPHA448).
The clinical characteristics of both types of haemophilia are similar: spontaneous or traumatic haemorrhages; muscle haematomas; haemophilic arthropathy caused by recurrent bleeding into target joints; and bleeding into the CNS. Without suitable exogenous clotting factor replacement therapy these manifestations of the disease could result in disabling or even deadly sequelae, which negatively impact patients’ quality of life and reduce their life expectancy.2
Until the 1960s, haemophiliac patients were treated with blood or plasma to compensate for their deficit in clotting factors. The low plasma concentration of coagulation factors made it necessary to administer large volumes of replacement factors with the consequent problems derived from hypervolaemia. Subsequently, the advent of more refined techniques for purifying these proteins made it possible to obtain purer and more concentrated products, with a very notable reduction in the quantities to be injected and in the levels of contaminating proteins, which often resulted in anaphylactic reactions. As a result, patients’ quality of life was significantly improved. These plasma proteins, isolated by immunoprecipitation techniques with monoclonal antibodies, were first developed in 1982 and are still being used for the treatment of haemophilia A and B.2
The landmark event that opened the door to the current recombinant treatment of haemophilia was the isolation and cloning of the genes that produce clotting factor VIII (1984) and clotting factor IX (1985).2 From that moment onwards, recombinant technology made it possible to prepare replacement factors from mammalian and human cells, rather than human plasma, avoiding the notorious infections by emerging pathogens that had caused so much havoc to HIV patients in the 1980s.
At present, patients with haemophilia benefit from optimised treatment protocols based on systemic intravenous administration of clotting factors, either prophylactically or on demand. Conventional treatment is currently based on either plasma derived products, duly treated with heat and detergent to inactivate lipid coated viruses, or high purity recombinant coagulation factor concentrates devoid of human or animal proteins.3 4 There is almost universal consensus that recombinant products should be the treatment of choice.5
Infections with emerging pathogens (HIV, hepatitis C virus, prions, etc) in patients with haemophilia6–10 have contributed towards making these recombinant products universally available, as these products are highly efficient and offer high safety levels against such infections. In addition, they allow for an easy and effective dosing strategy as a result of their efficient control of in vivo recovery and a longer half-life,11 which has also made haemophiliac patients eligible for several invasive surgical procedures.12 13
Most patients with severe and hereditary haemorrhagic disorders were unable to benefit from the dental procedures they required, which often resulted in delays in the diagnosis and management of their dental conditions. Barriers to proper dental care were multifactorial and attributable to either the patient or the physician.
A case in point is that of adults older than 45 years. During their childhood or adolescence, these patients could benefit from neither plasma derived nor recombinant products. As a result, in their middle-age, they cannot help fearing that the currently available procedures could result in an increased bleeding risk, which more often than not has led to the neglect of dental care and to high proportions of untreated conditions, typically causing pain and resulting in complications following invasive dental procedures, such as root canal treatment or extractions.14
This paper reports on the case of a patient with severe haemophilia with a very good response to antihaemophilic treatment, who presented with mesial decay in the radicular and infragingival area of the upper left third molar as well as marginal gingivitis and tartar build-up in interproximal areas. Extraction of the involved tooth and scaling were indicated. Treatments based on recombinant factor concentrates have played a decisive role in improving the quality of life of haemophiliac patients by providing them with safe and efficient access to invasive surgical procedures, and particularly dental procedures.
Case report
The patient was a 60-year-old Caucasian man with severe type A haemophilia (<1% factor VIII) who presented to the dental clinic with pain in the upper left third molar (tooth No 28). The tooth was buccally displaced with food impaction on its mesial surface. There was decay in the mesial radicular and infragingival areas, which made extraction of the tooth advisable. He experienced a throbbing sensitivity to cold, which took a few seconds to subside, and exhibited marginal gingivitis and tartar build-up in interproximal areas, particularly on the lingual aspect of the lower incisors. Scaling was indicated.
He had no deleterious habits but reported a family history of type 1 and 2 diabetes mellitus. He had moderate hypertension, treated with 20 mg of enalapril maleate every 24 hours, and dyslipidaemia, treated with 5 mg of rosuvastatin every 24 hours. Laboratory results were as follows: glucose 88 mEq/L; aspartate transaminase 46 IU/L; alanine transaminase 52 IU/L; gamma-glutamyltransferase 141 IU/L; bilirubin 0.58 mg/dL; haemoglobin 17.5 g/dL; creatinine 1.22 mg/dL; platelets 418 000/μL; CD4 528/μL; high density lipoprotein 33 mg/dL; low density lipoprotein 103 mg/dL; cholesterol 163 mg/dL; triglycerides 132 mg/dL; glomerular filtration rate 63 mL/min; and serum vitamin D deficiency 18 ng/mL (treated with 266 μg hydroferol every 15 days).
He was being treated on demand with 50 IU/kg of ADVATE recombinant factor VIII (Baxalta Belgium Manufacturing SA). ADVATE (octocog alfa human coagulation factor VIII) is synthesised in Chinese hamster ovary cells using recombinant DNA technology. As a third generation recombinant factor, no exogenous human or animal derived proteins are used in the cell culture, purification, or final formulation processes. The product is indicated for intravenous administration.
The number of units administered is expressed in international units (IUs), the WHO’s standard for factor VIII products. Factor VIII activity in plasma may be expressed either as a percentage (relative to normal human plasma) or in IUs (relative to the international standard for factor VIII in plasma); 1 IU of factor VIII activity corresponds to the amount of factor VIII present in 1 mL of normal human plasma, which means that the required dose of factor VIII must be determined using the following formula: required IUs=body weight (kg)×desired factor VIII rise (%)×0.5. This formula is based on the principle that 1 IU of factor VIII per kg of body weight increases plasma factor VIII activity by 2 IU/dL. Evaluation of the immunogenicity and type of ‘inhibitor kinetics’ induced by ADVATE is carried out by calculating the reduction in clotting factor activity in a mixture of an exogenous source of the coagulation factor (eg, normal pooled plasma) and potentially inhibitor positive plasma. The Nijmegen assay15 is a modification of the Bethesda assay developed by Kasper et al.16 This modified method has been recommended by the Factor VIII/IX Scientific Subcommittee for FVIII Inhibitor Testing of the International Society of Thrombosis and Haemostasis17; 1 Nijmegen-Bethesda unit (NBU) is defined as the amount of inhibitor that produces 50% residual factor VIII activity. A positive inhibitor titre is defined as >1 BU/mL by the Bethesda method and by >0.3 NBU/mL by the modified Bethesda method.
Patient preparation
Three hours before his dental appointment, the patient was administered 50 IU/kg of ADVATE recombinant factor VIII, which would ensure 100% plasma concentration of the deficient clotting factor. All dental procedures described here were performed in a private clinic. Both patient and physician signed an informed consent form.
Haematological and coagulation tests
Once the patient signed the required informed consent form, blood samples were placed into tubes containing 0.105 M sodium citrate in a 1:10 ratio (V/V). Plasma was obtained following centrifuging at 2000×g for 20 min. All haemostasis related parameters, such as prothrombin time (PT), prothrombin activity, activated partial thromboplastin time (aPTT) (cephalin time), and fibrinogen, were calculated in accordance with established standards. The HemosIL reagent kit (Instrumentation Laboratory; Bedford, Massachusetts, USA) was used according to the manufacturer’s instructions. The HemosIL RecombiPlasTin 2G kit was used to measure PT and fibrinogen levels, as described by Tripodi.18 The HemosIL APTT-SP (liquid) reagent was used to determine the value of aPTT, as described by Van de Bresselaar.19 20
The Coamatic (Chromogenix: Instrumentation Laboratory) chromogenic assay was used to determine factor VIII concentration and activity levels.21 This method is based on the notion that factor X is activated to factor Xa by factor IXa in the presence of calcium and phospholipids. The reaction is catalysed by factor VIII, which may be considered a cofactor. If enough Ca2+ and phospholipids are used together with significant levels of factors IXa and X, activation of factor X will only depend on the amount of factor VIII that is present. Factor Xa hydrolyses the S-2765 chromogenic substrate and releases the pNA chromophore, producing a yellowish solution that absorbs light at a wavelength of 405 nm. The intensity of the factor Xa generated (reflected by the colour intensity of the solution) is proportional to the factor VIII activity of the sample. Addition of the I-2581 synthetic thrombin inhibitor prevents the generated thrombin from hydrolysing the S-2765 substrate. Reagents and substrates were reconstituted following the manufacturer’s instructions.
Plasma samples were diluted in a HEPES buffered saline solution (20 mM HEPES, 145 mM NaCl, pH 7.4) containing 1% bovine serum albumin in 10 IU/mL. The result was subjected to a 10-fold dilution in factor VIII deficient plasma (Siemens Healthcare Diagnostic Products GmbH, Marburg, Germany) containing 1 IU/mL of von Willebrand factor. For each analytical set, a phial of the WHO’s eighth IS calibrator was equilibrated to room temperature for 15 min before reconstitution in 1 mL Milli-Q H2O. After reconstitution, the calibrator was kept for an additional 15 min at room temperature and was transferred to Nunc-Immuno Tubes (Minisorp; Thermo Fischer Scientific, Waltham, Massachusetts, USA), where it was stored on ice for a maximum of 3 hours. The samples and the calibrator were further diluted at 20 mIU/mL in diluents from either the kits or bovine serum albumin. Following the manufacturer’s instructions, several dilutions were subsequently prepared. Calibrator/samples were mixed with reagents containing factor X, factor IXa, (pro)thrombin, phospholipids, and calcium, and incubated for a specific time period after which reagent containing factor X was added. All reactions were performed at 37°C and absorbance at 405 nm was measured continuously on a SpectraMax Microplate Reader (Molecular Devices, Sunnyvale, California, USA) for 5 min. SoftMax Pro 5.4.1 software (Molecular Devices) was used to determine the increase in absorbance over time.
Clinical dental procedure
All routine and specific dental procedures were carried out with the utmost care and meticulousness. Scaling was performed using a Satelec ultrasonic probe with a No 1 tip, at minimum power and with abundant irrigation. Following the removal of deposits, teeth were polished using a low speed contra-angle handpiece and a rubber cup with Detartrine abrasive paste. For the extraction, following a rinse with 2% chlorhexidine, a topical anaesthetic gel was applied in both the buccal and palatal areas. A lidocaine carpule with 0.012% epinephrine was infiltrated into the buccal area as anaesthesia. Palatal anaesthesia was also applied. The syndesmotomy was performed using a straight root elevator, which was also employed for a slight preliminary dislocation. The extraction was performed using upper molar forceps, chiefly with palatal–buccal and circular movements. After the avulsion, a curette was used to ensure that no inflammatory tissue remained in the tooth socket. A dressing was applied to the wound so that the patient would compress it when closing the mouth.
Postoperatively, the patient was advised to compress the cotton for at least 1 hour. He was warned against any movement that could dislodge the clot and increase bleeding. Spitting, rinsing, toothbrushing, and eating or drinking hot foods or beverages were all contraindicated for at least 24 hours.
The postoperative antihaemophilic treatment regimen was established at 50 IU/kg of factor VIII at 12 hours and subsequently every 24 hours for the next 2 days.
Global health problem list
One of the most relevant breakthroughs in the history of scientific knowledge was the discovery of DNA by Watson and Crick in 1953. This enabled certain proteins to be prepared by recombinant techniques for their therapeutic use in clinical practice. Hundreds of recombinant therapeutic proteins have been produced to date by means of the expression of a therapeutic gene from a fragment of DNA and have become the therapy of choice for many diseases.
Clotting factors VIII and IX are two such proteins. The two chief advantages of recombinant drugs are their high efficacy and safety against pathogen derived infections. Their pharmacokinetic profile is a major boon to their ease of use as their well controlled in vivo recovery and longer half-life allow for a straightforward and efficient dosing strategy.
Although recombinant products are associated with a very favourable pharmacokinetic profile, the behaviour of the drug is highly dependent on the characteristics of each patient, especially as far as in vivo recovery and half-life are concerned. The half-life of factor VIII is around 12 hours, although there may be a certain degree of interpatient variability. For that reason, it is essential to evaluate the drug’s effects in each patient at least once a year in order to establish the optimal dose. In this study, the so-called chromogenic or colorimetric analysis was used to determine factor VIII plasma levels and conduct the required pharmacokinetic analysis.21 22 This is the method of choice for diagnosing haemophilia A or B and to establish the severity of the disease.
The baseline (pre-factor VIII treatment) coagulation profile of the patient was as follows: PT 12.6 s (reference value 8–13 s); prothrombin activity 82.7% (reference value 75–120%); aPTT (cephalin time) 58.7 s (reference value 24–37 s), and fibrinogen 318 mg/dL (reference value 150–450 mg/dL). As was to be expected, only the aPTT value was deviant. The patient’s inhibitor titre was below 0.3 NBU/mL.
Figure 1 shows a Bayesian estimation of the patient’s pharmacokinetic parameters. Factor VIII levels recovered to 81.9 UI/dL at 3 hours and to 22.0 UI/dL at 24 hours from infusion. Clearance was 0.028 dL/h/kg; volume of distribution at steady state was 0.5 dL/kg; time to 1% above baseline was 89.0 hours; and half-life was 14.5 hours.

Bayesian estimation of the patient’s pharmacokinetic parameters. The curve illustrates factor VIII activity over time following intravenous administration of factor VIII. The grey area represents the deviation (±20%) with respect to the general population; (- - -), pharmacokinetics in the general population; (♦), patient’s data; (---), Bayesian pharmacokinetic estimation.
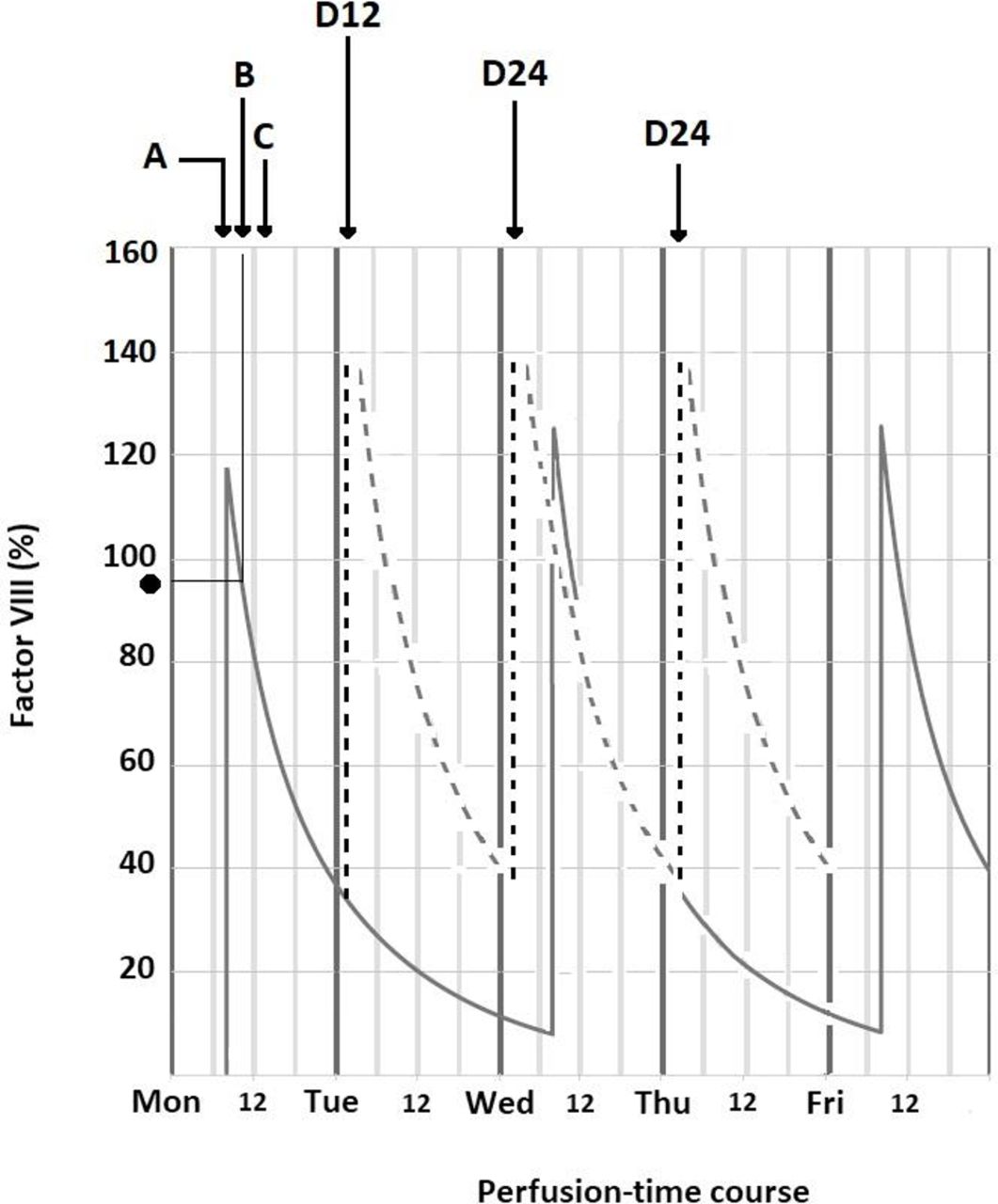
Figure 2 shows how the dose was optimised. Arrow A indicates pretreatment factor VIII administration; arrow B indicates the length of the dental procedure; arrow C, the onset of the recovery period; and arrow D administration of the prophylactic treatment. At the time of the procedure, factor VIII plasma levels were higher than 90%, while factor VIII levels remained between 40% and 140% throughout the recovery period.
{kind=link}
{kind=link}
Dose optimisation. Prediction of the patient’s factor VIII plasma levels. (A) Pre-procedure factor VIII administration. (B) Dental procedure. (C) Onset of recovery period. (D) Prophylactic treatment: 12 hours (D12) and 24 hours (D24). (·) Factor VIII plasma level (>90%) at the time of the procedure. Factor VIII remained between 40% and 140% (- - -) during the recovery period.
The lack of a clear definition of what constitutes major or minor surgery in the context of haemophilia leads to inconsistencies in the management of these patients23 given the fact that there is considerable variability in the categorisation of major and minor surgical procedures and a certain overlap in surgical nomenclature. Even so, most authors admit that dental surgical procedures such as those reported in this study should be considered major surgery when performed in patients with severe haemophilia.24–28 Although the patient in this study was a severe haemophiliac and was subjected to a major surgical procedure, the same methodology applied to patients with no bleeding disorders was used. If a thorough haematological investigation is performed, adequate levels of factor VIII are maintained, and clotting time is properly monitored, there is no reason why routine techniques cannot be used both for scaling procedures and extractions. As a precaution, post-extraction compression of the surgical site should be maintained for longer than in non-haemophiliac patients. Use of tranexamic acid in the gauze used for compression and during rinsing in the next few days is not indispensable but can be extremely useful as an emergency treatment in case the haemorrhage proves impossible to control with simple compression. In our case, the patient’s response to antihaemophilic therapy was satisfactory, which precluded the need for any emergency measures. Clotting times were within the ranges expected in a severe haemophiliac with factor cover. In other cases, the patient’s poor response or the highly invasive nature of the procedure may complicate the recovery process. In our patient, postoperative bleeding was within the expected range: intense during the extraction procedure but gradually decreasing after that. The patient was instructed to present to the dental clinic and the haemophilia unit for follow-up 1 month after the operation.
Global health problem analysis
If health education and, particularly, an awareness of the importance of dental hygiene and regular dental check-ups are essential to avoid dental disease in the general population, they are even more crucial in patients suffering from coagulopathies in order to avoid the complications associated to a higher bleeding risk. Haematologists and dentists should assist patients and their families in identifying the resources they can avail themselves of to benefit from adequate dental care. A close relationship between haematologists, dentists, oral surgeons, and paediatric surgeons is as essential as undergoing annual dental check-ups, as oral health is an essential component of overall health.29
As a chronic disease caused by a deficiency in the proteins responsible for blood clotting, haemophilia results in sequelae affecting mainly the musculoskeletal and nervous systems. It may also be associated with multisystem involvement in patients with HIV or hepatitis C virus infection. For that reason, the approach to these patients should be multidisciplinary.30 During the planning phase, it must be ascertained that the procedure is carried out without complications. The surgical team must be trained to manage unforeseen situations and make any necessary changes to postoperative protocols.
Under this multidisciplinary perspective, in order to help prevent and manage postoperative bleeding in haemophiliac patients, the dentist must contact the haematologist before the operation to explain the dental procedure he wishes to undertake, report on the patient’s medical condition, and describe the kinds of local postoperative haemostatic measures he intends to apply.31 The haematologist will then prescribe the most appropriate antihaemophilic treatment with sufficient anticipation to ensure that plasma levels of the deficient clotting factor are optimal during the procedure.
Specifically, it is recommended31 that factor VIII should remain between 50% and 75% of normal levels prior to oral and periodontal surgery, and between 75% and 100% before maxillofacial surgery. Before an anterior alveolar nerve block, lingual infiltration or floor of mouth injection, replacement therapy with an exogenous coagulation factor is required. Restorative, prosthodontic, endodontic, and orthodontic treatments are considered safe for most patients with bleeding disorders. The dentist must immediately inform the haematologist about any cases of prolonged bleeding, dysphasia, or speaking or breathing difficulties following a dental procedure.
In order to facilitate and optimise dental procedures in patients with haemophilia, a tool has recently been developed that is capable of quantifying and managing the risk of a dental haemorrhage. The purpose is to avoid unnecessary treatments and promote multidisciplinary collaboration between the different clinical teams in attendance.14 The tool takes into account the perception of the invasiveness of the dental procedure and the severity of haemophilia (mild, moderate, severe) to stratify patients into four groups: no bleeding risk group, low risk group, moderate risk group, and high risk group, establishing a connection between the invasiveness of the dental treatment, the bleeding risk, and the required haemostatic cover.
The past decade has seen a burgeoning of innovative treatment options for haemophilia,32 33 ranging from recombinant next generation clotting factors with longer half-lives, to a wide range of new approaches and modalities, such as the new subcutaneously administered bispecific antibody mimicking factor VIII that binds to activated factor IX, which in turn activates factor X. Other new developments include liver targeted siRNA, which can result in a large enough knockdown of the antithrombin gene to allow significant haemostasis, and an antibody against a tissue factor pathway inhibitor that also enhances haemostasis.34
This new therapeutic scenario, described by some authors as ‘miraculous’,35 has resulted in improved standards of care for persons with haemophilia. The improvement has allowed a breaking down of the barriers that prevented these individuals from benefiting from dental procedures, which resulted in delays in the diagnosis and management of their dental conditions. More often than not, these barriers were related to fears: the fears experienced by dentists, who lacked the experience needed to manage these kinds of patients, and those experienced by patients, especially older patients, who still remembered the times when any procedure, even a minor one, entailed a high bleeding risk. These fears led to a situation whereby dental care in this group of patients was often neglected, with a high number of dental conditions being left untreated, which caused pain and added complexity to the procedures, particularly extractions, when they were performed. As a result, a high proportion of patients with bleeding disorders required continuous dental care, often because of their few (if any) visits to the dental clinic,14 with high rates of decay and gingival disease (83% and 46.7%, respectively).
This new therapeutic context has made it possible to use conventional techniques in almost all kinds of surgical procedures without increasing the risk of bleeding.
These achievements have been possible as a result of the development of recombinant coagulation factors, which have become the treatment of choice for the disease. They are highly efficient and boast an excellent safety profile. Moreover, their well controlled in vivo recovery and longer half-life allow for a straightforward and efficient dosing strategy. Given their excellent safety profile against emerging pathogens, they can be used at the correct dose without fearing potential contamination.8
A third aspect to be considered with respect to the new protocols used for the treatment of haemophilia is the significant increase observed in patient adherence to regular dental check-ups, which used to be practically null.
Finally, the current situation could improve even further when advanced therapies, in both their cell and gene based modalities, now still at a budding stage, become established as ‘curative’ protocols that replace the current (merely palliative) treatments of the disease.36
The recombinant era has revolutionised therapeutic options in terms of safety and efficacy. It has also opened up the possibility to offer patients personalised and individualised treatments.
As far as haemophilia is concerned, patients with the disease currently benefit from safe and highly efficient treatment protocols based on recombinant factors. DNA technology based recombinant factors are currently displacing human plasma derived clotting factors, which in the 1980s gave rise to one of the worst disasters in the history of medicine when several thousand people were administered HIV and hepatitis C virus contaminated products. This happened because, in spite of being asymptomatic, many of the blood donors recruited to achieve therapeutically effective factor concentrates had a significant yet undetectable emerging pathogen transmission potential. This caused thousands of patients to die and made many others avoid the use of plasma derived products for fear of contagion. All of this resulted in many patients receiving suboptimal treatment and made it unfeasible to implement certain treatment protocols, particularly surgical ones, prompting the development of arthropathic sequelae and dental conditions, among others.
Prepared without human plasma components, recombinant factors have become the treatment of choice for haemophilia as their favourable safety profile protects them against viral and prionic agents. The most recent recombinant factors are particularly efficient as they are obtained from human cells, which significantly reduces their immunogenicity, decreases patients’ immune responses, and lowers the levels of anti-factor antibodies.37
These new developments have allowed a more robust dose optimisation given both the greater efficacy of the products used and the longer half-life associated with the new coagulation factors. This has also impacted the way haemophiliac patients are managed in dental practice. The above mentioned pharmacological benefits of the new products, combined with appropriate healthcare education and medical specialisation programmes addressed to patients and their families on the one hand, and to dental professionals on the other, have minimised the risks associated with dental procedures and improved the dental health of patients of all ages.38
As a result, currently, there are no significant dental health differences between child and adolescent haemophiliac patients and their non-haemophiliac counterparts. Differences do however exist in the adult patient population given the sequelae haemophiliac adult patients have had to put up with as a result of not having benefited from optimal treatments during their childhood and adolescence.39
Patient’s perspective
As a 60-year-old patient with severe type A haemophilia who has participated in this study, I have to say that my childhood and youth were very hard because there were no treatments with coagulation factors. Bleeds were recurrent and joint damage turned into severe haemophiliac arthropathy with joint sequelae, which have now become disabling. In addition, mild or more invasive surgical interventions were almost unthinkable for patients like myself.
The current therapeutic scenario for haemophilia has made it possible to improve patient management and quality of life. It has also made it possible to break down the barriers that prevented people like me from benefiting from standard treatments. Nowadays, I can safely undergo treatments for dental disease with much less risk of bleeding, thanks particularly to the advent of the new recombinant factors, which have shown themselves to be safer and more effective. Indirectly, this has led to increased adherence to prevention protocols and periodic reviews.
Learning points
In this case report, the patient’s response to anti-haemophiliac therapy was satisfactory, which precluded the need for any emergency measures.
Clotting times were within the ranges expected in a severe haemophiliac with factor cover. Postoperative bleeding was within the expected range: intense during the extraction procedure but gradually decreasing after that.
The past decade has seen a burgeoning of innovative treatment options for haemophilia, which have resulted in improved standards of care for persons with haemophilia.
The improvement has allowed the use of conventional techniques in almost all kinds of surgical procedures without resulting in a higher risk of bleeding, increased observed patient adherence to regular dental check-ups, and a better patient quality of life.
References
Footnotes
Contributors AL and LR conceived and designed the study. AL performed the formal haematological analysis. LR performed the clinical dental procedure. AL wrote the manuscript.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Patient consent for publication Obtained.