Article Text

Abstract
Rhabdomyolysis is a serious and life-threatening condition which has many established causes including endocrine disturbances. Of those, thyroid, adrenal and pituitary deficiencies are the most commonly seen. Most cases of rhabdomyolysis with adrenal insufficiency that have been reported have been primary. Here, we report an encounter with a patient who presented with her second case of severe rhabdomyolysis in the setting of secondary adrenal insufficiency. The cause for corticotropic suppression was most likely autoimmune hypophysitis given the presence of other autoimmune comorbidities including a new diagnosis of autoimmune hepatitis. In addition to her case, we present a brief review of the literature pertaining to cases of rhabdomyolysis attributed to adrenal insufficiency.
- adrenal disorders
- pituitary disorders
- thyroid disease
- muscle disease
- drugs: musculoskeletal and joint diseases
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
- adrenal disorders
- pituitary disorders
- thyroid disease
- muscle disease
- drugs: musculoskeletal and joint diseases
Background
This case represents an unusual presentation of rhabdomyolysis in secondary adrenal insufficiency as well as multiple autoimmune conditions occurring in the same patient. It highlights the importance of screening for endocrinopathies, in particular cortisol deficiency, in patients who present with rhabdomyolysis and an unclear precipitant. It is one of only few reported cases of secondary adrenal insufficiency causing rhabdomyolysis and thus represents an important clinical learning point. It also highlights the challenges and limitations in investigating the causes of hypopituitarism, and in particular, the diagnosis of autoimmune hypophysitis.
Case presentation
A 55-year-old woman was admitted to a tertiary hospital with a 4-day history of profound global weakness, light-headedness and cola-coloured urine following some moderate exertion which involved packing to move house. She was unable to walk or even lift her head off of the bed. Her swallow and respiratory effort were also impaired. She denied any preceding symptoms suggestive of an infection. She also denied any alcohol intake.
Her medical history was significant for a previous episode of uncomplicated rhabdomyolysis 9 years prior. At that time, the peak creatine kinase (CK) was 9672 U/L and she was admitted for a short duration in a rural hospital. No obvious precipitant was found at that time and the only hormonal parameter tested was thyroid-stimulating hormone (TSH) which was normal. One month after that episode, her thyroid function tests were repeated and she was diagnosed with autoimmune hypothyroidism with a TSH of 5.3 mIU/L (reference 0.5–4.0), free thyroxine of 9 pmol/L (reference 10–25) with the detection of antithyroid peroxidase antibodies (238 IU/mL [reference <50]). She was commenced on levothyroxine. Her other comorbidities included well-controlled depression, anxiety and chronic degenerative back pain for which she was on long-term methadone. Her regular medications included thyroxine 75 µg, sertraline 200 mg and methadone 50 mg daily. She was a non-drinker, had never smoked and denied a history of illicit drug use.
Investigations
Biochemical parameters for the current admission are shown in table 1. She was found to have severe rhabdomyolysis with a CK of 62 270 U/L. Screening bloods on presentation also revealed markedly elevated transaminases. Although this transaminase rise is likely due to her skeletal muscle damage, raised gamma-glutamyl transferase, alkaline phosphatase and bilirubin levels prompted us to screen for liver diseases. Her complete blood exam, renal function and electrolytes were normal.
Laboratory parameters for the current case
A screen for causes for rhabdomyolysis was undertaken. Myositis immunoblot was negative except for a positive Ro-52 which is not specific for autoimmune myositis. A muscle biopsy was not done given the paucity of other clinical signs to suggest an inflammatory myopathy. She had an elevated total IgG level, and in particular IgG1 was markedly raised (table 1). A screen for endocrinopathies is shown in table 1. Her thyroid function tests on presentation showed that she had slightly high TSH with thyroxine (T4) which was in the lower end of the normal range suggesting that she was under-replaced and her dose of levothyroxine was subsequently increased to 100 µg. A morning cortisol was 9 nmol/L, indicating profound adrenal insufficiency. A similar result was found on a repeat testing. The paucity of symptoms of adrenal insufficiency in this case was surprising and suggestive of a chronic process. A morning adrenocorticotropic hormone (ACTH) level was undetectable, indicating that the cortisol deficiency was secondary to hypopituitarism. The remainder of the pituitary screen revealed a suppressed follicle-stimulating hormone (FSH) and luteinizing hormone (LH) which strengthens the diagnosis, although there was sparing of the other axes. An MRI pituitary was unremarkable.
On further investigating the cause of the deranged liver function tests, an ultrasound abdomen showed a nodular heterogeneous liver, moderate ascites and moderate splenomegaly, suggestive of hepatic cirrhosis with portal hypertension. A screen for cause of liver disease revealed positive anti-soluble liver antigen (anti-SLA) antibodies and raised IgG suggestive of autoimmune hepatitis. All other liver autoantibodies were negative and the remainder of the screen did not reveal an alternate cause for the deranged liver function tests.
Treatment
She was initially managed with intravenous hydration, general supportive measures and oral hydrocortisone replacement. Her clinical condition improved.
Outcome and follow-up
Her CK rapidly dropped over the first week and returned to the normal range within 3 months (figure 1). She was able to walk within 2 weeks and was discharged with rehabilitation in the home at this time. A liver biopsy several months into her treatment with hydrocortisone replacement found features of cirrhosis and inflammation that would be in keeping with, but not specific for, a partially treated autoimmune hepatitis.

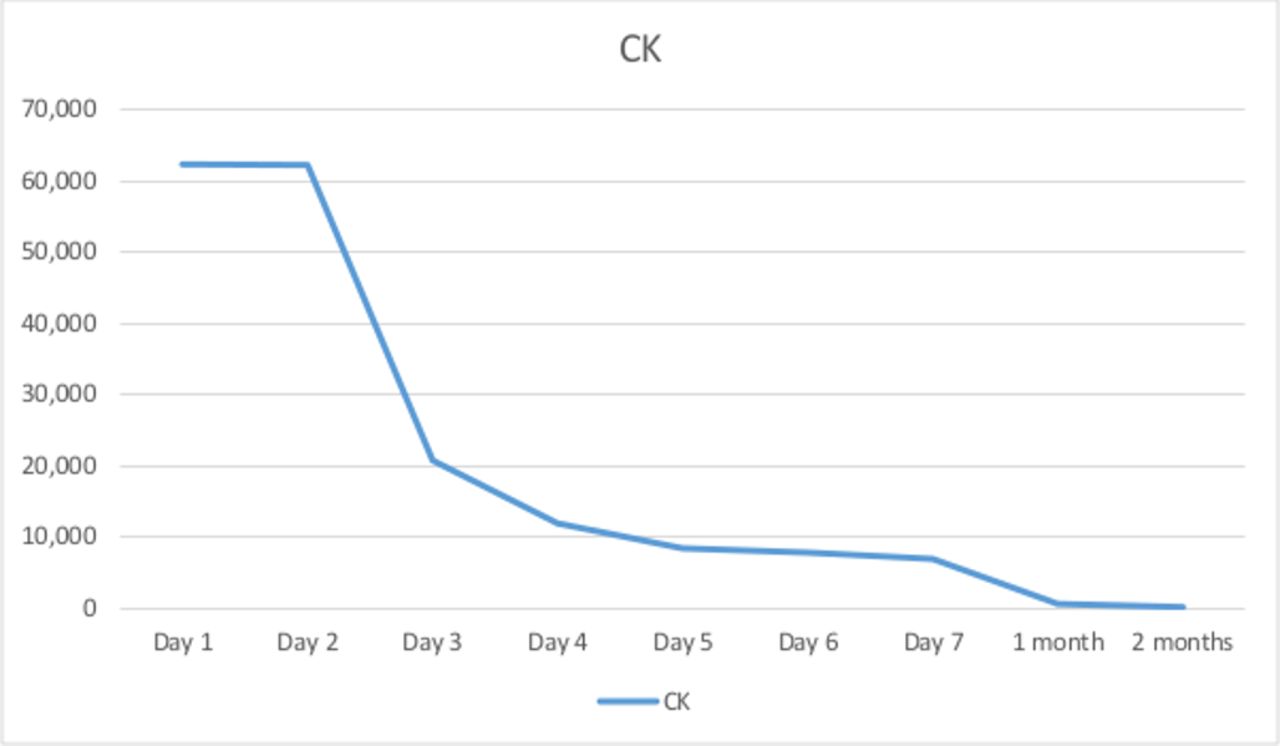{kind=link}
Trend in CK during week 1 and 2 months following presentation. CK, creatine kinase.
Discussion
Rhabdomyolysis can be a life-threatening condition with a mortality rate as high as 59% reported in critically ill patients with acute kidney injury (AKI).1 2 Clinical definitions vary, however, generally a serum CK of at least 10 times the upper limit of normal is considered diagnostic, particularly when followed by a fairly rapid descent to normal levels.3 The classical clinical presentation includes weakness, myalgia and pigmented urine, although this triad is seen in <10% of patients. With severe rhabdomyolysis, the most critical complication is AKI which is thought to be caused by myoglobin obstructing the tubules causing acute tubular necrosis.3 Other serious complications include metabolic disturbances and compartment syndrome. Causes for rhabdomyolysis are vast and include trauma, drugs, seizures, neuromuscular disorders, myopathies and hormonal abnormalities among others.3–5
Rhabdomyolysis in the setting of adrenal insufficiency is rarely seen. There are 12 other cases described in the literature, 7 being primary adrenal insufficiency alone, 1 being primary adrenal insufficiency with concurrent hypothyroidism and the remaining 4 found to be secondary to hypopituitarism. The characteristics of these cases are seen in table 2. Most of the cases described were associated with hyponatremia, which alone has also been implicated as a cause for rhabdomyolysis. This understandably led some authors to speculate that it may play a key role in the mechanism of rhabdomyolysis in adrenal insufficiency.6 However, the current case and several others show a markedly raised CK without hyponatremia which strongly points towards an alternative mechanism by which adrenal insufficiency leads to skeletal muscle breakdown. It certainly is known to present with a variety of other musculoskeletal complications including flexion contractures, myopathies, back pain, hyperkalemic neuromyopathy, Guillain-Barre syndrome, migratory myopathy and sciatica-like pain.7–9 It may be that cortisol deficiency, similar to the proposed mechanism in hypothyroidism,10–12 leads to muscle breakdown by impaired glycogenolysis or impaired mitochondrial oxidative metabolism. Another possibility is that the hypotension or shock associated with adrenal insufficiency may lead to muscle ischaemia.
Characteristics of previously reported cases of rhabdomyolysis with primary or secondary adrenal insufficiency
With regards to the reported cases of rhabdomyolysis in the setting of both hypocortisolism and hypothyroidism, it is unclear which deficiency played the larger role in inducing muscle damage as they were both treated concurrently with resolution of signs and symptoms. Certainly hypothyroidism has a strong association with myopathy and it is a known cause of rhabdomyolysis.4 However, in the current case, the patient was already on treatment for hypothyroidism. At a mild degree of under-replacement as indicated by her low-normal T4 and mildly raised TSH, one would not expect to see such a severe degree of rhabdomyolysis. Thus, the current case suggests that the rhabdomyolysis seen in hypopituitarism or concurrent deficiencies in both cortisol and thyroid hormone is caused, to a meaningful degree, by the hypocortisolism.
One of the interesting aspects of this case is the coinciding diagnosis of liver disease. Liver disease does have an association with adrenal insufficiency through a number of mechanisms. For instance, hepatic cirrhosis has an established association with adrenal insufficiency via suppression of the hypothalamic–pituitary–adrenal axis,13 and conversely there are also documented cases of liver cirrhosis thought to be caused by hypopituitarism via changes akin to the development of metabolic syndrome leading to non-alcoholic fatty liver disease.14 The latter mechanism is a possibility in this case; however, the presence of anti-SLA antibodies and hypergammaglobulinaemia points more towards the diagnosis of an autoimmune hepatitis and she did not have clinical features of metabolic syndrome. The diagnosis of autoimmune liver disease in a patient with an established diagnosis of chronic autoimmune thyroiditis does then raise the suspicion of a predilection for autoimmune conditions as is seen in autoimmune polyglandular syndrome (APS). There are recorded case studies of autoimmune hepatitis in all three categories of APS.15–19 This has implications for screening for other autoimmune conditions and in this case raises the suspicion of an autoimmune cause for the adrenal insufficiency.
With regards to the profound chronic adrenal insufficiency in this case, the finding of low ACTH as well as low FSH and LH was diagnostic for a central endocrine pathology. Hypopituitarism has a number of causes including mass lesions, infiltrative disease, infarction, radiation, infection and autoimmunity. The hormonal deficiencies seen are highly variable, ranging from the classical panhypopituitarism to the loss of only one or two tropic axes.20 In the current case, in which there is a loss of the corticotropic and gonadotropic axes, the presence of concurrent autoimmune conditions and the paucity of radiological findings or features suggestive of an alternative cause, strongly points towards an autoimmune aetiology, that is, autoimmune hypophysitis.
The term autoimmune hypophysitis encompasses a spectrum of histopathological entities including granulomatous, lymphocytic and xanthomatous forms.21 Lymphocytic hypophysitis is the most common form.21 Histologically it is characterised by inflammation and cellular infiltration of the pituitary gland.22 It can present with symptoms of mass effect such as headache and diplopia, or with variable disturbances of the hypothalamic–pituitary axis.21 23 The most commonly affected axes include corticotropic, thyrotropic, lactotropic and gonadotropic.22 24While the precise pathogenesis of this condition is unknown, it has been associated with pregnancy and also certain immunological therapies.21 It also has an association with other autoimmune conditions, including autoimmune hepatitis,25 and has been seen in cases of APS.26MRI of the pituitary can be normal but is more often abnormal. Typical findings include thickening of the pituitary stalk with or without extension to the basal hypothalamus, diffuse enlargement of the gland which may resemble an adenoma, absence of bright spot of the posterior lobe on T1-weighted images, contrast enhancement, symmetrical sella floor and sellar mass.24 The availability and clinical utility of anti-pituitary antibodies (APAs) are evolving, but at present they are still predominantly restricted to the domain of clinical research.24 Presently, the limitations for clinical use include inconsistent methodology, low sensitivity and specificity.24 They can be positive in normal subjects as well as other pituitary conditions such as Sheehan syndrome and pituitary adenoma.24 In the current case, the MRI was non-diagnostic and there were no APA assays available for clinical use in the state. However, given the concurrent autoimmune diagnoses, the pattern of deficiency and the paucity of evidence for another cause for hypopituitarism, autoimmune hypophysitis is the most likely diagnosis.
A delayed presentation of Sheehan syndrome was another diagnosis considered in this case. Sheehan syndrome is hypopituitarism resulting from ischaemia of the pituitary gland occurring at the time of childbirth usually where there has been postpartum haemorrhage and hypotension.27 It often presents acutely following birth with an Addisonian crisis but can also present subacutely with the first sign being an inability to lactate.28 There are also cases where the diagnosis has been made many years following birth when they have presented with chronic hypopituitarism.28 We found one such case in the literature in which the patient initially presented with rhabdomyolysis (table 3).29 She was found to be deficient in all of the anterior pituitary hormones and gave a history from many years ago of having had postpartum haemorrhage plus an inability to lactate following birth.29 There is another report of a late diagnosis of Sheehan syndrome causing acute renal failure but with only a mildly elevated CK (table 3).30 The renal failure was thought to be related to chronically low cortisol, and in that case they had elicited no history of peripartum complications, only an inability to breastfeed.30 In the current case, the patient did give birth over 20 years prior but did not report any complications and had been able to breastfeed. One could entertain a possibility of a variant of Sheehan syndrome with sparing of the lactotropic axis, and certainly in the current case, the prolactin level was in the normal range. However, while Sheehan syndrome cannot be entirely excluded, it is less likely than autoimmune hypophysitis.
Anterior pituitary parameters in reported cases of rhabdomyolysis or acute renal failure with secondary adrenal insufficiency
Hypopituitarism secondary to chronic methadone use is another possibility but is also less likely in this case. Most of the described cases are opiate-related hypogonadotropic hypogonadism rather than a deficiency in other anterior pituitary hormones.31 Opiate-induced secondary adrenal insufficiency is thought to be uncommon with only very few case reports in the literature but this may be due to under-reporting.32–37 Some believe that the prevalence of opiate-induced adrenal insufficiency due to suppression of hypothalamic–pituitary–adrenal suppression could be as high as 9%–29%.38 In the current case, as discussed above, the presence of other autoimmune tendency would favour an autoimmune hypophysitis over secondary adrenal insufficiency from opiate use although this cannot be entirely excluded. Also, as far we know, there is no known association with therapeutic opiate use and rhabdomyolysis. Most reported cases of rhabdomyolysis that have been described are in the setting of an opiate overdose resulting in immobilisation.39 40
There are isolated case reports of rhabdomyolysis in association with selective serotonin reuptake inhibitors (SSRIs) and in particularly with sertraline use.41 42 Almost all reported cases were seen within 4 months after commencing or uptitrating an SSRI and in the setting of strenuous exercise.41 43 Few other cases have also been reported with SSRI overdose.44 45 Our patient was on sertraline for several years, and there were no recent dose changes or a history of overdose. She also did not engage in strenuous exercise. Although a contribution from sertraline to rhabdomyolysis cannot be ruled out, it is less likely in our patient. We also could not find any clinical cases of adrenal insufficiency in association with SSRI use.
This dilemma highlights the need for improved diagnostic tools for hypopituitarism. Both the rarity and the wide spectrum of clinical manifestations of autoimmune hypophysitis make it difficult to achieve consistency in diagnosis. As discussed, contrast MRI and serum APAs may help to a limited degree. Histological confirmation by transsphenoidal biopsy can lead to a more definitive diagnosis and is the gold standard, but is usually only obtained in cases in which the mass effect is causing symptoms such as headache and visual disturbance and in these cases often surgery is being considered.46 Differentiating hypophysitis from tumour would be beneficial in these instances as the former may then be managed with corticosteroids, avoiding surgery and thus preserving remaining pituitary function.
Learning points
If the cause of rhabdomyolysis is unclear after an initial workup and preliminary investigations, one should actively screen for cortisol deficiency.
Although most recognised cases of adrenal insufficiency in association with rhabdomyolysis are primary, there seems to be an association with secondary adrenal insufficiency. The pathogenesis seems to be due to an autoimmune process.
Once rhabdomyolysis in association with cortisol deficiency is established, screening for other autoimmune diseases should be considered depending on the clinical suspicion and biochemical abnormalities.
References
Footnotes
LK and SN contributed equally.
Contributor LK and SN contributed equally.
Contributors LK wrote parts of the case presentation, discussion and drafted the tables for the manuscript. LK also consented the patient for publication. SN initiated the write-up. He also wrote several parts of the discussion, edited the manuscript several times and inserted a number of references. He also answered all the reviewer comments. Both authors have contributed equally to this manuscript.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Patient consent for publication Obtained.