Article Text

Abstract
Hyponatraemia is a common electrolyte disturbance with multiple causes. We present a case of a 49-year-old Caucasian female with cholangiocarcinoma, who had a hyponatraemia which was initially assumed to be based on a syndrome of inappropriate antidiuretic hormone secretion as paraneoplastic phenomenon. At physical examination, hyperpigmentation was seen and multiple episodes with syncope were reported. Subsequent endocrine assessment with a synthetic adrenocorticotropin hormone (ACTH) stimulation test and measurement of ACTH levels revealed primary adrenal insufficiency also known as Morbus Addison. We started hydrocortisone and fludrocortisone replacement therapy, resulting in resolving of symptoms, hyponatraemia and hyperpigmentation.
- adrenal disorders
- pancreatic cancer
- thyroiditis
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Background
Hyponatraemia is a common clinical problem with a multifactorial aetiology. The general incidence of hyponatraemia (serum sodium <136 mEq/L) in hospitalised patients is 30% and in 2.6% serum sodium is <124 mEq/L.1 One of the causes of hyponatraemia is hypocortisolism. Early recognition of this disease is of vital importance, because hypocortisolism is a potentially life threatening condition and presenting symptoms are often non-specific.
The first diagnostic steps in hyponatraemia are to confirm a hypo-osmolar hyponatraemia using serum osmolality, this measurement should match the calculated osmolality. Urinary osmolality is also necessary and measures ADH activity, which is present when urinary osmolality ≥100 mOs/kg. Urinary sodium can help determine if sodium loss is renal of non-renal. The use of diuretics should always be considered, as it makes urinary sodium and osmolality more difficult to interpret.
After confirming a hypotonic (serum osmolality <275 mOsm/kg) hyponatraemia, in absence of ADH activity, the differential diagnosis includes primary polydipsia, tea and toast and beer drinkers hyponatraemia. If urinary osmolality ≥100 mOs/kg, urinary sodium and assessment of a patients’ volume status are needed.2
A hyponatraemia with a low urinary sodium (<30 mmol/L) and a hypovolaemic state can be the result of vomiting, diarrhoea, sequestration of fluid and recently stopped diuretics. If the patient is hypervolaemic, cardiac failure, liver cirrhosis and nephrotic syndrome needs to be considered.
Hyponatraemia with a high urinary sodium (≥30 mmol/L) and a hypovolaemic state can also be the result of vomiting, but also the use of diuretics or primary adrenal insufficiency. If the patient is euvolaemic secondary adrenal insufficiency and SIADH need to be considered.
In case of hyponatraemia without hypotonicity, other conditions should be considered. This can be the result of a hyperglycaemia or exogenous solute (ie, mannitol, intravenous immune globulines). In these situations, water is drawn from intracellular to the extracellular space, resulting in a lowered serum sodium concentration, but hypertonicity. Another consideration is a pseudohyponatraemia, which is a laboratory miscalculation of the serum sodium. This phenomena can be hyperlipidaemia, hyperproteinaemia or obstructive jaundice. The electrolyte concentration is measured in plasma, which normally consists of about 93 percent water and seven percent solids, mostly proteins and fats. The serum sodium concentration is determined based on the measured plasma sodium concentration and the estimated percentage of water. An increase in solids leads to a lower water percentage, and therefore a miscalculation of the serum sodium, however tonicity remains normal.2
In this article, we present a case of hyponatraemia and hyperpigmentation caused by a Morbus Addison in a patient with jaundice caused by a pancreatic carcinoma. Initially, it was interpreted as SIADH, but hyperpigmentation was the vital clue in diagnosing primary hypocortisolism.
Case presentation
A 49-year-old Caucasian woman with a medical history of auto-immune hyperthyroidism was hospitalised for analysis of painless jaundice under suspicion of a cholangiocarcinoma. At presentation, she had a hypotonic hyponatraemia (serum sodium 124 mmol/L, measured serum osmolality 255 mOsm/kg, calculated serum osmolality 260.3 mOsm/kg) combined with a urine osmolality of 678 mOsm/kg and a urine sodium of 219 mmol/L. Awaiting further diagnostics (eg, morning serum cortisol, thyroid-stimulating hormone and thyroxine) she was initially treated for the assumed diagnosis of a syndrome of inappropriate antidiuretic hormone secretion (SIADH) as paraneoplastic effect of a suspected cholangiocarcinoma. However, fluid restriction failed to raise the serum sodium level. In addition, on further inquiry our patient reported multiple episodes of syncope in the previous weeks. Closer physical examination showed a severe hyperpigmentation, which our patient reported to be worsened over the last year (figure 1). Furthermore, our patient is 177 cm tall and has a body weight of 55.15 kg with a BMI of 17.6 kg/m2. The combination of severe hyperpigmentation, hyponatraemia and episodes of syncope and orthostatic hypotension (with blood pressure values between 73/50 mmHg and 90/52 mmHg and a heart rate of 75/minute) made hypocortisolism caused by primary adrenal insufficiency, also known as Morbus Addison, the most likely diagnosis.
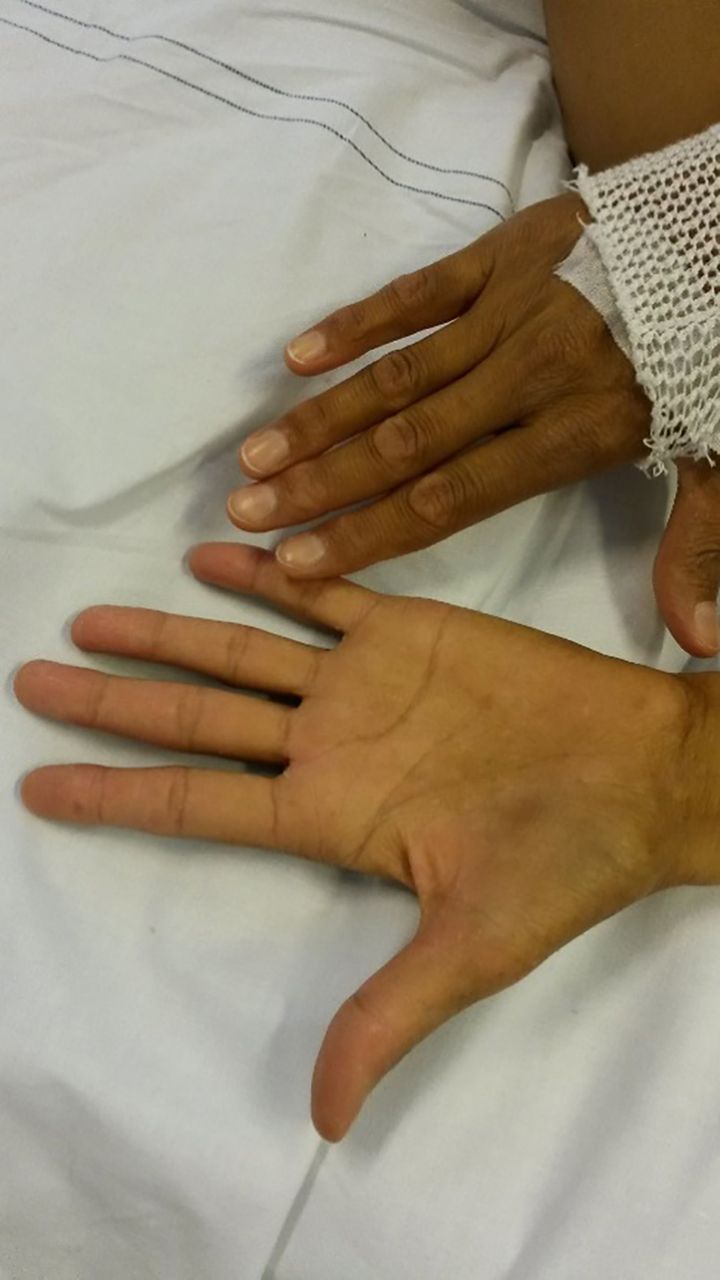
Skin hyperpigmentation of a Caucasian female.
Investigations
To confirm our hypothesis of primary adrenal insufficiency, we performed a synthetic adrenocorticotropin hormone (ACTH) stimulation test (synacthen 250 µg) showing a cortisol decrease from 217 nmol/L to 207 nmol/L at 30 minutes. Basal plasma ACTH level was 492 pmol/L (normal range 0–11 pmol/L) and adrenal auto-antibodies were positive. Serum renin concentration was 1520 µU/mL (normal range 5–60 µU/mL) whereas serum aldosteron was not detectable (<50 pmol/L). These results confirmed the diagnosis of Morbus Addison. Abdominal CT-scan, performed in the diagnostic process of the suspected cholangiocarcinoma, showed no abnormalities of the adrenal glands.
Differential diagnosis
Our patient had a hypotonic hyponatraemia (serum sodium 124 mmol/L, serum osmolality 255 mOsm/kg, calculated serum osmolality 260.3 mOsm/kg) combined with a urinary osmolality of 678 mOsm/kg and a urinary sodium of 219 mmol/L. Pseudohyponatraemia, for example as a result of jaundice, was excluded as cause because the hyponatraemia was hypotonic hyponatraemia.
She was initially assessed as euvolaemic, leading to a differential diagnosis of SIADH. In daily practice, SIADH is often assumed to be the cause of hyponatraemia, even before the diagnostic work-up is completed. This was also the case in our patient, in whom treatment for SIADH was initiated before the adrenal insufficiency was adequately ruled out. Recent guidelines also state that a SIADH diagnosis requires exclusion of other possible causes of hyponatraemia, like adrenal insufficiency.3 In our case, the assumed diagnosis SIADH resulted in delay in the final diagnosis.
Treatment
Our patient had both an auto-immune hyperthyroidism and a Morbus Addison and was diagnosed with a polyglandular autoimmune syndrome type 2.4 We started cortisol replacement treatment with hydrocortisone 50 mg intravenous once, followed by oral hydrocortisone 20 mg–10mg–10mg per day. The dose was doubled because of simultaneous use of rifampicine. We also started fludrocortisone 62.5 µg per day. This treatment was indicated because in our patient, there was a primary cause of adrenal insufficiency. Therefore, mineral corticosteroid is also lacking and substitution is essential.
Outcome and follow-up
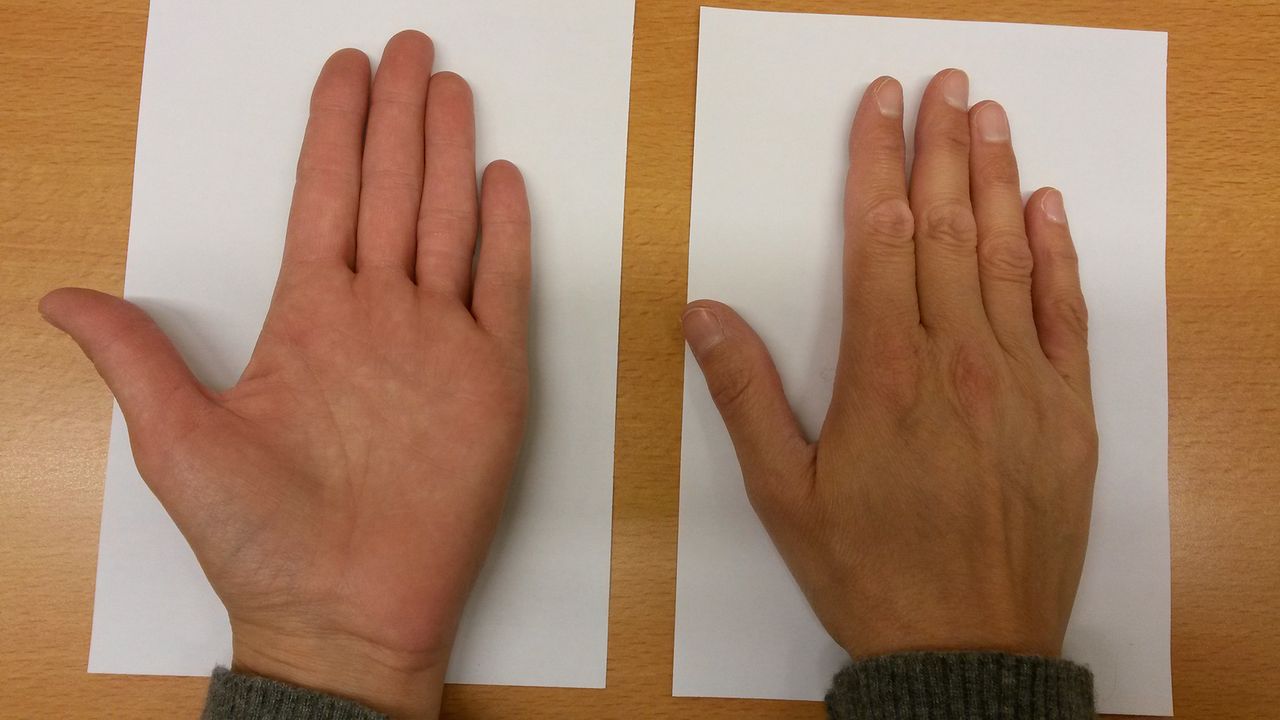
Our patient was treated for her adrenal insufficiency resulting in full recovery of symptoms and a normalisation of hyponatraemia in 4 days. Also, within 2 weeks the hyperpigmentation resolved (figure 2).
{kind=link}
{kind=link}
Normal skin colour after hydrocortisone treatment.
Analysis for the jaundice of our patient initially led to the suspicion of cholangiocarcinoma, but histological examination revealed pancreatic adenocarcinoma. She underwent a pancreaticoduodenectomy. The operation was, unfortunately, complicated by intra-abdominal bile acid leakage treated with a temporary percutaneous biliairy stent. Unfortunately, during oncological follow-up metastasis and later also local recurrence of the pancreatic carcinoma were diagnosed, therefore palliative chemotherapy was given, despite this effort our patient deceased 2 years after diagnosis.
Discussion
In this article, we present a case of hyponatraemia and hyperpigmentation caused by Morbus Addison in a patient with jaundice as a result of pancreatic carcinoma. Morbus Addison is a rare disease, and pancreatic carcinoma occurring simultaneously is, to our knowledge, never reported before. It did result in an inappropriate initial diagnosis, as SIADH is a common finding in patients with malignancies. We did find a case of a woman with autoimmune polyglandular syndrome type 2 having the combination of hyponatraemia, hyperpigmentation caused by Morbus Addison with a previous autoimmune Hashimoto’s thyroiditis.5 In our case, undersubstitution of levothyroxine could have contributed to the hyponatraemia, however, thyroid hormone concentration was normal with a levothyroxine dose of 100 µg/day.
Hyponatraemia in patients with hypocortisolism is due to aldosterone deficiency, leading to renal sodium loss, and is enhanced by water retention via an increased release of antidiuretic hormone (ADH) in response to a reduction in systemic blood pressure and cardiac output caused by cortisol deficiency. The ADH response is maintained by cortisol deficiency because the direct negative feedback of cortisol on ADH release is declined.6 This explains why in hypocortisolism hyponatraemia does not improve with only fluid restriction.
Hyperpigmentation in primary adrenal insufficiency is caused by an increased production of α-melanocyte-stimulating-hormone (αMSH).7 Both αMSH and ACTH originate from the pro-hormone peptide pro-opiomelanocortin (POMC). In primary adrenal insufficiency POMC production is strongly increased in response to the fall in cortisol levels with concomitant release of αMSH causing a bronze hyperpigmentation.
Early recognition of Morbus Addison is of vital importance, because hypocortisolism can be a life threatening condition in case of physical stress as activation of the hypothalamic-pituitary-adrenal axis is an essential response during illness. Furthermore, Morbus Addison often presents with non-specific symptoms, which makes hyperpigmentation an important clue in the diagnosis.4
Learning points
Hyponatraemia can be the first presentation of Morbus Addison.
In patients with hyponatraemia and co-morbidity predisposing to SIADH, adrenal insufficiency should always be considered as contributory factor or primary cause.
Hyperpigmentation caused by increased production of α-melanocyte-stimulating-hormone can be a vital clue to consider primary hypocortisolism, also when other serious co-morbidity is present.
Hyperpigmentation in this case of Morbus Addison resolved within 2 weeks after starting cortisol replacement therapy.
Footnotes
Contributors The manuscript has been approved by all authors and has never been published, is not under consideration in the similar form in any other peer-reviewed media. To the best of our knowledge, all aspects of our work has been written with accuracy and no conflict of interest, financial or other, exists. Detailed author contributorship statement: BJMB; corresponding author, initiated the case for a case report together with JA. Together with JA and RAF, the design of the paper was established. BJMB conducted and revised the paper, generated laboratory results and patient information data from our patient information system, made pictures of the hands and collected patients approval. JA; initiated the case for a case report together with BJMB. Together with BJMB and RAF, the design of the paper was established. Alsma provided analysis and interpretation of data, revised different concepts of the paper critically and has given his final approve before publishing. RAF; established together with BJMB and JA, the design of the paper. RAF provided analysis and interpretation of data, revised different concepts of the paper critically and has given his final approve before publishing.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.
Patient consent for publication Obtained.