Article Text

Abstract
A 44-year-old Caucasian man with a history of deceased donor renal transplant for end-stage renal disease from Alport’s syndrome (AS), presented with a spontaneous subarachnoid haemorrhage and hydrocephalus. Following an external ventricular drain for the hydrocephalus, a CT angiography revealed a dissection of the left vertebral artery extending into vertebro-basilar junction necessitating a bypass between left occipital artery to left posterior inferior cerebellar artery. He had a posterior fossa Craniectomy, C1 laminectomy and coiling off, of the left vertebral artery. Postprocedure course was prolonged but uneventful with complete recovery and normal renal function 18 months postpresentation. AS, a disease caused by abnormalities in the synthesis of type IV collagen, can cause aneurysms with severe and permanent neurological sequalae. We present a case of AS with intracranial arterial dissection with potential life-threatening consequences and discuss the genetic and molecular basis of AS along with review of the relevant literature.
- neuroimaging
- neuro itu
- interventional radiology
- renal transplantation
- stroke
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Background
Alport’s syndrome (AS) is a rare genetic disorder and typically presents with nephropathy secondary to progressive glomerulonephritis, high tone sensorineural deafness and ocular abnormalities (anterior lenticonus, peri macular flecks, spontaneous capsular rupture).
The pathophysiology in AS is explained by mutations in the genes encoding alpha-3 (IV), alpha-4 (IV), or alpha-5 (IV) chains of type IV collagen found in the basement membranes. Collagen forms the main building blocks of the arterial walls, and there is a strong molecular basis behind occurrence of major arterial aneurysm in disorders of collagen synthesis. However, presentation of patients with AS with intracranial aneurysms is minimally studied and reported.
This case highlights the need for awareness of this rare, but potentially life-threatening complication in patients with AS. Large-scale evaluation of the surgical and autopsy specimens of vascular aneurysms in patients with collagenopathy is needed to establish the relation better. Also, future clinical and epidemiological studies are warranted to emphasise stronger and well-defined association between vascular and intracranial aneurysms in patients with collagen synthesis dysfunction such as in AS.
Case presentation
A 44-year-old Caucasian man transferred to the neurosurgical unit following a spontaneous subarachnoid haemorrhage with hydrocephalus. He initially presented to a local hospital with a 24-hour history of sudden, severe headache with associated nausea and vomiting. On arrival, his Glasgow Coma Scale (GCS) was 15/15. Following multiple episodes of intravenous morphine for pain, he went into respiratory distress requiring ventilatory support. Subsequently, he transferred to our facility for higher level of care.
Patient’s prior medical history included end-stage renal disease (ESRD) from AS, 20 years ago. He was on haemodialysis for 8 years, subsequently received a diseased donor renal transplant and was on Tacrolimus and Mycophenolate for immunosuppression. His other past medical history included, non-hodgkin’s lymphoma treated with chemo and radiation and in remission. He was also on aspirin, furosemide, ranitidine, senna, atenolol, calcium supplements and paracetamol. He was a non-smoker, and his maternal uncle had ESRD from AS.
Soon after his admission to the neurosurgical unit, he received an external ventricular drain for the hydrocephalus. Laboratory work-up revealed, stable serum creatinine at his baseline, normal blood urea nitrogen, electrolytes, complete blood count and urinalysis. A CT angiography (CTA) revealed a dissection in the intradural segment of the left vertebral artery (figure 1), which continued distally beyond the origin of the left posterior inferior cerebellar artery (PICA) on to the vertebro-basilar junction.

CT Angiogram with a dissection in the intradural segment of the left vertebral artery.
Investigations
CTA of the brain.
Differential diagnosis
Acute ischaemic or haemorrhagic cerebrovascular accident.
Treatment
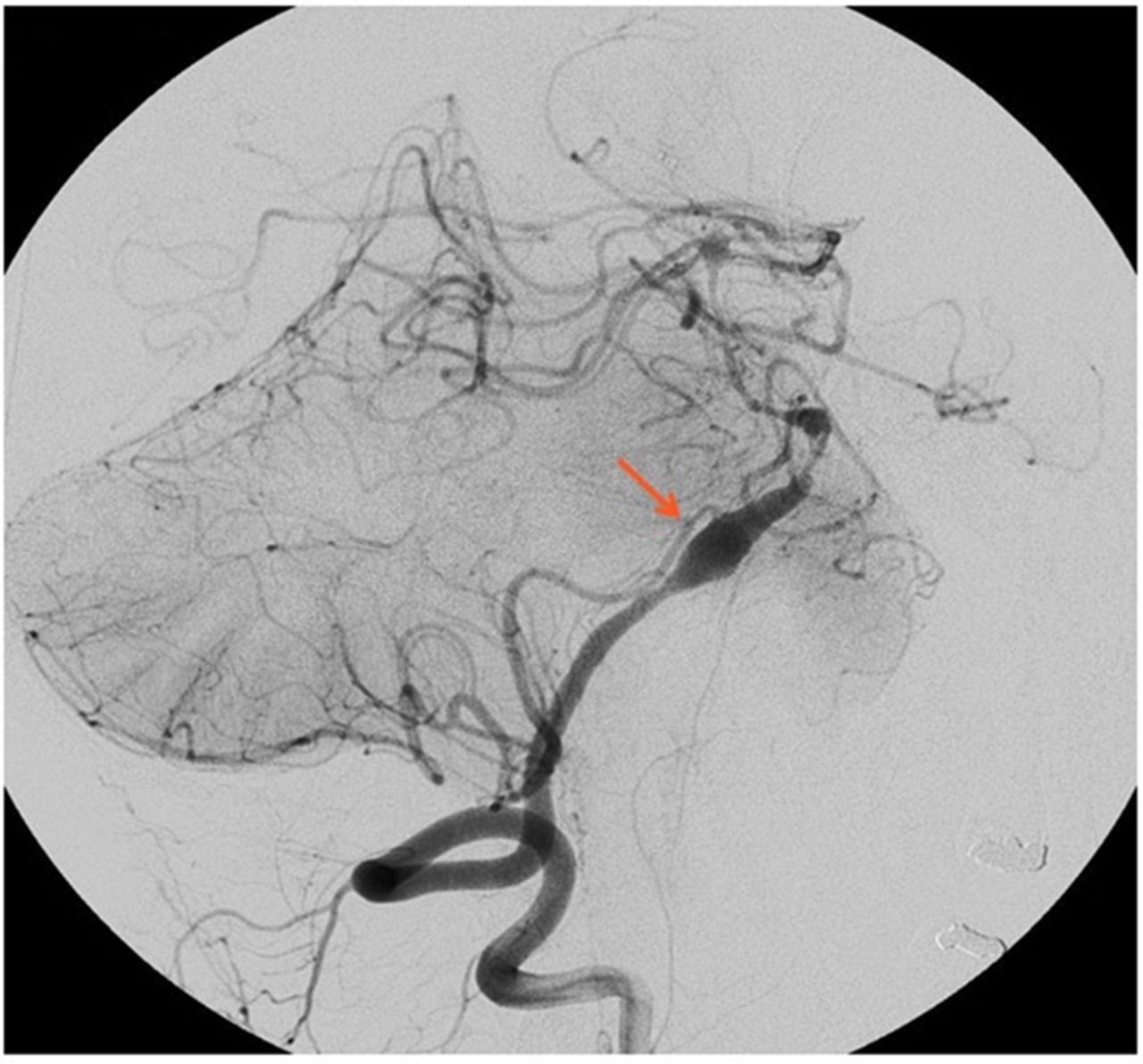
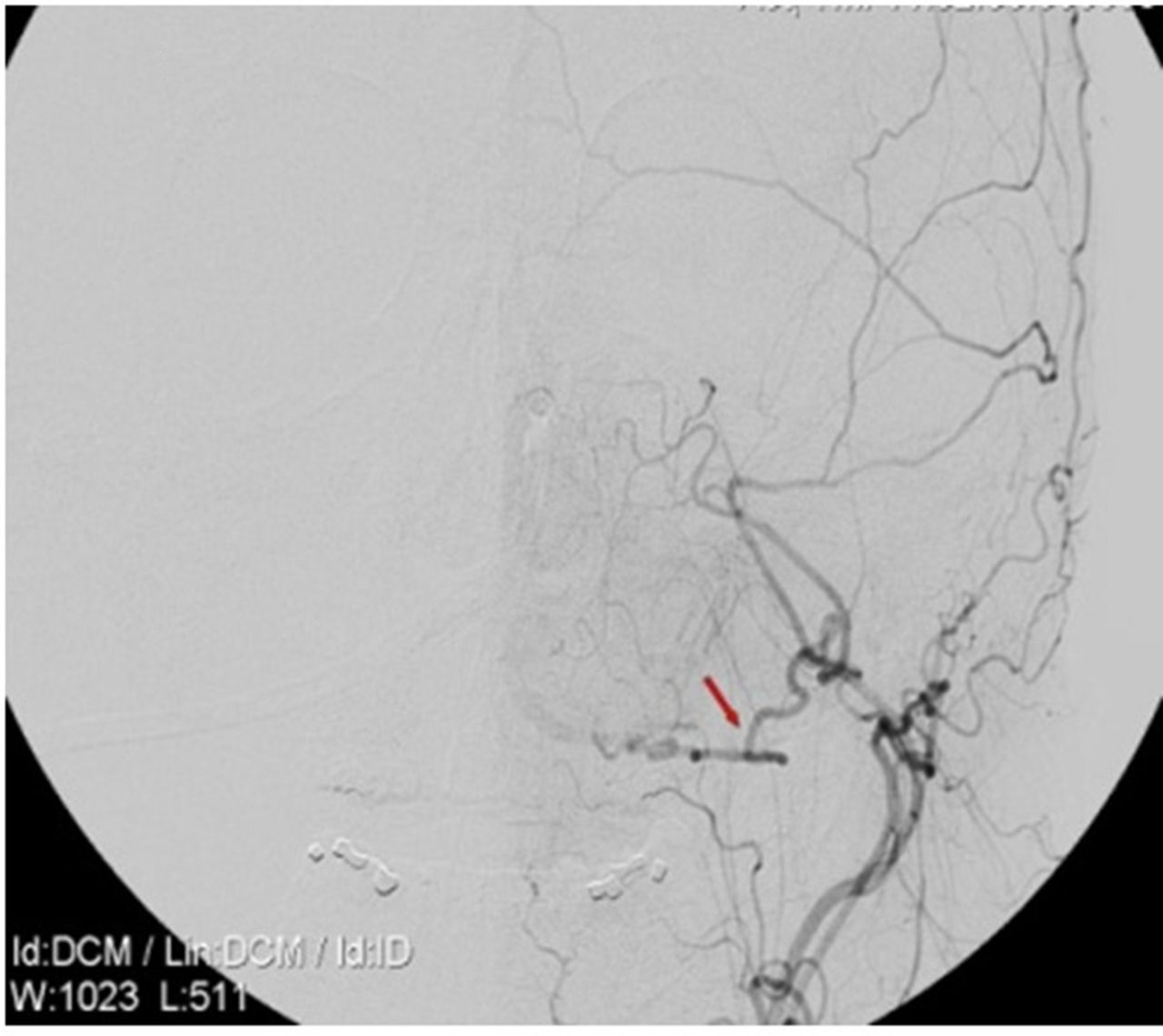
He underwent left occipital artery to PICA bypass (figure 2) along with a standard posterior fossa craniectomy with C1 laminectomy. Postbypass the left vertebral artery was coiled off (figure 3). The occipital artery and PICA bypass was patent. The angiogram for the procedure, however, revealed a dissection of the right vertebral as well with possible involvement of the basilar artery. Given the high possibility of causing infarction with further intervention on the only remaining vertebral vessel, decided to treat conservatively. His post procedure imaging revealed possible area of ischaemia in the brain stem and the cerebellum (PICA territory). He had a prolonged period of recovery in the neuro intensive care unit and recovered to a GCS of 14/15 (E4V4M6). A programmable ventriculo-peritoneal shunt was inserted and discharged to the local neuro rehabilitation unit in a stable condition.

Left occipital artery to posterior inferior cerebellar artery bypass.

{kind=link}
{kind=link}
{kind=link}
Left vertebral artery coiled off.
Outcome and follow-up
Eighteen months following his presentation, he remained symptom free with complete recovery, and his renal function remained at baseline.
Discussion
In 1927, an English Physician, Cecil A Alport described members of three generations in a family, with combinations of progressive hereditary nephritis and hearing impairment.1 Subsequently, many more families were described with similar characteristics, and the eponymous title, ‘Alport’s syndrome’ was coined in 1961.2 Though incidence is unknown, AS has been implicated in renal replacement therapy for ESRD in 1.5% of children in Europe.3 Data from the USA demonstrates that AS accounts for 3% of children with ESRD and 0.2% of adults with ESRD.4
Molecular biology and genetics of AS
AS is genetically heterogeneous, and it results from mutations in one of the three subunits of a type IV collagen subunits (alpha chains), that makes a type IV collagen molecule, which forms a significant component of the basement membranes. The three sub units of the type IV collagen molecule, the alpha (IV) chains, are intertwined into a triple helical structure. Two molecules interact at the C-terminal end, and four molecules communicate at the N-terminal end to form a ‘chicken wire’ network. Alpha (IV) chains have six isomers and are designated as alpha-1 to alpha-6. Normal glomerular basement membrane (GBM) contains two collagen networks, the first consisting of the alpha-1 (IV)/alpha-2 (IV) chains and the second of the alpha-3 (IV)/alpha-4 (IV)/alpha-5 (IV) chains.5 In, table 1, details of the various tissue distributions of Alpha (IV) chains of collagen are included.
Tissue distribution of alpha chains of type IV chains of collagen
Overall, the primary micro-structural abnormality in patients with AS results from abnormalities of basement membrane expression of alpha-3 (IV), alpha-4 (IV), and alpha-5 (IV) chains of type IV collagen. In the basement membrane of a patient with AS, these chains are either under expressed or absent. A spectrum of mutations in type IV collagen genes can lead to expression of different phenotypes and give rise to variable clinical presentations. Table 2 depicts the mutation of the genes coding for alpha chains of type IV collagen in AS.
Mutations of the genes coding for alpha chains of type IV collagen in AS
There are three genetic forms of AS that are recognised:
X-linked: The most common form (85%) that results from mutations in the encoding of the alpha-5 (IV) chain of type IV collagen.
Autosomal recessive: Caused by mutations in genes encoding either alpha-3 (IV) or alpha-4 (IV) chains and is responsible for approximately 10%–15% of cases.
Autosomal dominant: Rare form that is caused by mutations in genes encoding either alpha-3 (IV) or alpha-4 (IV) chains.
Pathogenesis of aneurysms in AS
Our literature search for the reported cases of vascular aneurysms in AS (keywords: ‘Alport’s,’ ‘Aneurysm’) revealed only three reports,6–8 and did not yield reports with vertebro-basilar artery aneurysms. The index patient has history of AS and had a deceased donor renal transplant for ESRD. He denied prior cerebro-vascular symptoms and hence did not receive vascular screening or angiogram before the presentation. Given the pathogenesis, we postulate that the genetic defect leading to abnormality in type IV collagen molecule may have resulted in the formation of the aneurysm in our patient with AS.
Yong-Zhong and van Alphen reviewed the major theories on the formation and growth of saccular aneurysms.9 Disruption of the internal elastic lamina is thought to be an critical requirement for the creation of saccular aneurysms, because it provides most of the strength to the cerebral arteries.9 Degeneration of this layer is a persistent feature of all saccular aneurysms. A coexisting medial defect and haemodynamic stress at an apex may aggravate degeneration at that point.10 11 Hypertension does not seem to be a significant originating factor in some aneurysm patients, but it may facilitate the formation and growth of saccular aneurysms,.11
Incidence of intracranial aneurysm in the children and young people may represent a distinct pathophysiological entity from adults.12 Cerebral, aneurysms in this age group fail to support a degenerative or chronic haemodynamic pathogenetic origin. Therefore, molecular and genetic aspect of ethology may have greater importance. Childhood intracranial aneurysms are relatively rare, and has been reported to have male predominance, predilection for posterior circulation and carotid bifurcation along with a propensity to reach larger sizes.13 14 Arterial deficiency of reticular fibres is of importance in the development and rupture of saccular aneurysm in childhood.15 We should, however, note that formation of premature vascular aneurysms and fragile nature of cerebral arteries commonly associated with type III collagen mutations.16 17
Specific connective tissue disorders such as Ehlers-Danlos syndrome, Marfan’s syndrome, osteogenesis imperfecta, pseudoxanthoma elasticum, neurofibromatosis type I and polycystic kidney disease, have been studied for their increased vascular fragility.18–20 Genetically defective form of type III collagen is believed to be a critical element in both vessel enlargement and aneurysmal rupture in these disorders.21 Fibromuscular dysplasia (FMD) is a multifocal, noninflammatory, nonatherosclerotic arteriopathy of uncertain aetiology, most commonly affecting segments of small to medium-sized arteries. Pathophysiological similarities between FMD and AS may be unclear, but there may be a common mechanism of pathogenesis in both syndromes, related to the expression of type IV collagen in the vascular basal membranes.22 23
Collagen Type IV and type V are localised to the endothelial basement membrane and basement membranes of smooth muscle cells of the intima and media.24 Type IV collagen plays a fundamental and vital role in vessel wall integrity, and it closely influences and interacts with various factors related to angiogenic activity.25 Skirgaudas et al studied expression and distribution of angiogenesis growth factors and selected vascular wall matrix proteins (fibronectin, Type IV collagen and alpha smooth muscle actin) in the walls of human intracranial aneurysms from surgical/autopsy specimen.26 Expression of matrix proteins on the aneurysmal vessel wall was found to be diffuse and disorganised as compared with their organisation in control vessels. A possible biological response related to the angiogenesis factors was noted as evident by strong patchy expression of vascular endothelial growth factors within the aneurysmal walls. Thus, abnormal or inadequate synthesis of type IV collagen subunits may possess a significant role in this pathological process.26
Our report raises the concern of vascular aneurysms in AS and stimulates the idea that it may be reasonable and advisable to look for collagenopathies in young patients with vascular aneurysms. ICA may be commoner in the population of collagenopathies than previously suspected and non-invasive screening may be appropriate. Little has been done to assess the risk of severe arterial aneurysms in AS and possible benefit of a screening protocol for eligible patients. Non-invasive low risk neuroimaging studies such as MR angiography (MRA) or CTA may prove to be useful screening modalities in selected cases. Large scale evaluation of surgical and autopsy specimens of vascular aneurysms from patients with known collagenopathy is needed to demonstrate more unambiguous molecular and genetic correlation of vascular aneurysms and collagen abnormalities. Further clinical and epidemiological studies are warranted to establish stronger and well-defined association between prevalence of intracranial and other vascular aneurysms in patients with collagen synthesis dysfunction such as in AS.
Learning points
Alport’s syndrome (AS) is a rare genetic disorder and typically presents with nephropathy from progressive glomerulonephritis, high tone sensorineural deafness, and ocular abnormalities.
AS is genetically heterogeneous, and results from mutations in one of the three subunits of a type IV collagen alpha chains, that makes a type IV collagen molecule.
Though rarely reported, vascular aneurysms may be commoner in the population of collagenopathies than previously suspected and non-invasive screening may be appropriate.
More extensive clinical and epidemiological studies are warranted to establish stronger and well-defined association between vascular aneurysms in patients with collagenopathies.
References
Footnotes
Contributors SB involved in the planning, conception, acquisition of data including images, patient consent, and NRA, SP and KMB helped with the final drafting of the case report. SB and NRA are the first authors.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Patient consent Obtained.
Provenance and peer review Not commissioned; externally peer reviewed.