Article Text

Statistics from Altmetric.com
Description
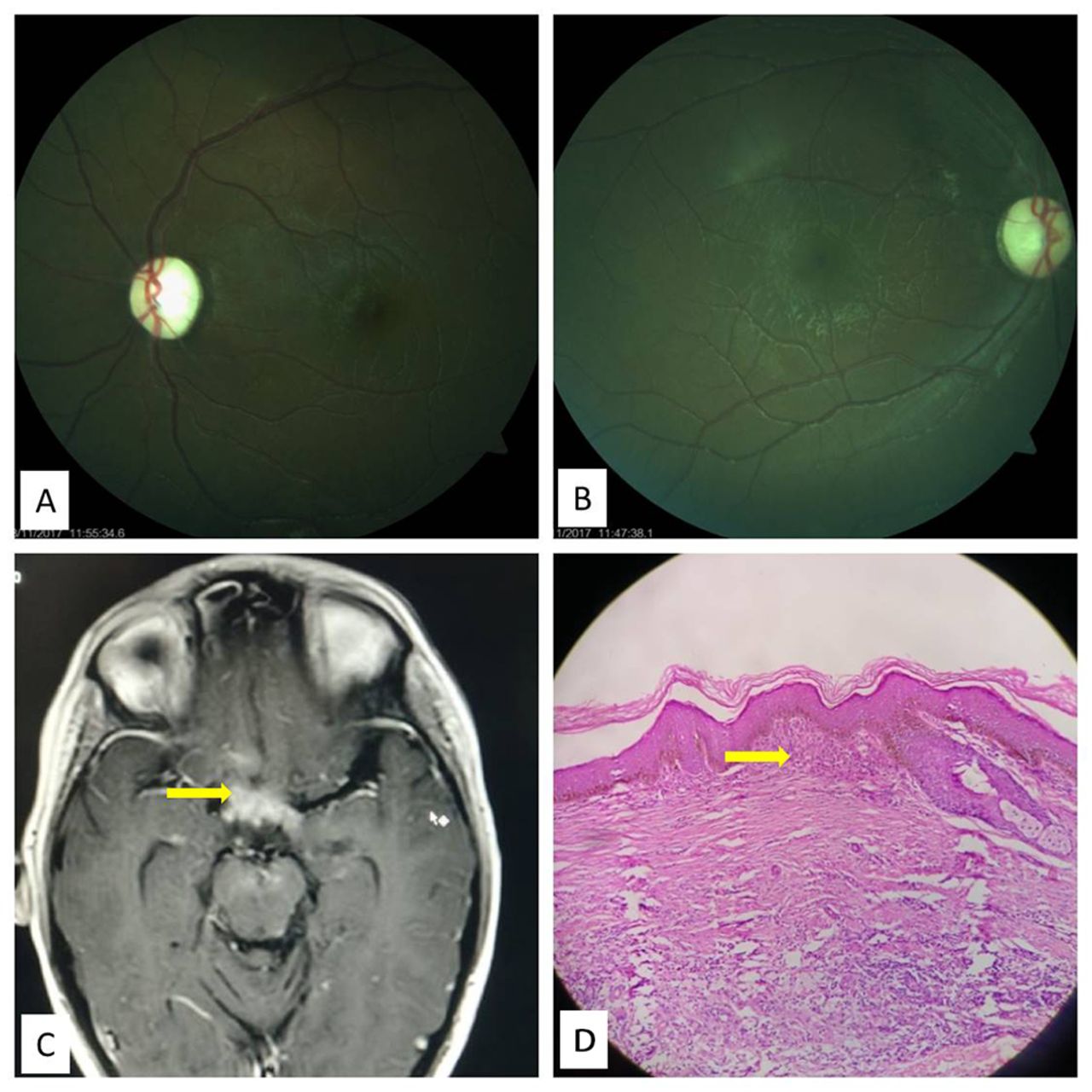
A 9-year-old male child presented with gradual progressive painless diminution of vision in both eyes since one and a half years. He had well-defined discrete hyperpigmented and hyperkeratotic papules of 3–5 mm in size involving right axilla, bilateral neck folds, nape of neck, waist, groin folds and popliteal fossa. A history of polyuria and excessive thirst was also noted. On ophthalmological examination, the best-corrected visual acuity was 20/100 in the right eye and counting fingers at 2 m in the left eye. The anterior segment was normal. Pupil light reflex was sluggish bilaterally. Fundus examination revealed symmetric total optic disc pallor with well-defined disc margins suggestive of optic atrophy (figure 1A,B). An MRI of brain and orbits was advised which revealed multiple T1 hypo-intense and T2/FLAIR hyper-enhancing intracranial lesions involving brain parenchyma, optic chiasma (figure 1C) and extending up to floor of frontal horn of lateral ventricle. Some local mass effect was noticeable without surrounding vasogenic oedema. A skin biopsy was performed which revealed diffuse histiocytic infiltration interspersed with mixed inflammatory cells, giant cells and foam cells (figure 1D). Immunohistochemistry was positive for CD68 (KP1) and negative for S100 protein. Hence a diagnosis of xanthoma disseminatum (XD) was made. The patient was treated with injection cladribine for 5 days following which there was a significant flattening of macula-papular lesions. At 3 months of follow-up, reduction in polyuria and excessive thirst were noted with supplementary oral desmopressin. Visual acuity remained stable.

{kind=link}
(A,B) Fundus photograph showing the bilateral optic atrophy. (C) Post-contrast-enhancing lesions visible around the pituitary region and optic chiasma. Flair MRI sequence showing hyper-intense intracranial lesions involving brain parenchyma and optic chiasma (arrow). (D) Histopathological examination showing basket weave keratosis with flattening of rete ridges in epidermis. A pandermal infiltration of histiocytic cells having foamy to pale eosinophilic cytoplasm with interspersed lymphocytes can be seen (arrow).
XD syndrome is a rare non-familial, normolipemic non-Langerhans cell histiocytosis. Males around 5–25 years of age are usually affected. Three clinical patterns have been suggested based on case reviews: persistent form (common) in which lesions usually never resolve, a rare self-healing form that is characterised by spontaneous resolution and a rare progressive form characterised by organ dysfunction and central nervous system involvement.1 Our patient belonged to the third category as evidenced by multiple intracranial lesions. Progressive form of XD is the rarest among the three forms as discussed. Our patient suffered from the progressive form with evidence of intracranial involvement. Literature reports mucous membrane involvement in patients with XD in around 40%–60% affecting oropharynx, larynx, cornea and conjunctiva. Literature has reported about maculo papular lesions around the eyelids and its margin, at corneo sclera junction. We could find only one case report where they had reported bilateral optic atrophy in male twins with XD, suggesting an association between them, though they could not find any infiltrative lesion on radioimaging.2 Our case had infiltrative intracranial lesions with involvement of the optic chiasma, thus explaining the bilateral optic atrophy with progressive form of XD, a very rare entity. Thus, our case report gives more prudent explanation for the bilateral optic atrophy which is seen with progressive form of XD. The classic triad of XD consists of cutaneous xanthomas, mucosal xanthomas and central diabetes insipidus due to infiltration of the xanthomatous cells in the hypothalamus-pituitary region. Diagnosis is mainly based on clinical and histopathological findings with CD68 positivity and negative S100, CD1a and Birbeck granules favouring the diagnosis of XD.3 Various treatment modalities have been tried, but the response to any form of treatment has not yielded very satisfactory results in most case reports.
Visual outcomes depend on the extent of involvement of the visual pathway. Drugs like vasopressin, chlorambucil, corticosteroid, azathioprine, cyclophosphamide, cladribine and vinblastine have been tried with variable response. Surgical excision and laser therapy have also been tried, but the course of the disease is characteristically punctuated with frequent relapses. The predilection of the xanthomatous cells for the hypothalamus-posterior pituitary region leading to central diabetes insipidus and the close anatomical relationship between the pituitary and optic chiasma can explain the spread of the lesions towards the chiasma, thereby resulting in bilateral optic atrophy.
Learning points
Early and appropriate neuroimaging modalities should be planned in cases with unexplained bilateral primary optic atrophy.
A multispeciality approach is required in cases of xanthoma disseminatum in order to understand the patho-mechanism and the various spectrum of the disease so that more definite and timely treatment modalities can be implemented.
Footnotes
Contributors MB,BT and NG contributed to diagnosis, workup, writing the manuscript and performing critical revision. PS holds the overall responsibility to the presentation and contributed in diagnosis and performing critical revision of the manuscript.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Patient consent Obtained.
Provenance and peer review Not commissioned; externally peer reviewed.