Article Text

Abstract
A 58-year-old man presented with a chief complaint of tongue indentations and discomfort. Otolaryngology treated him for oral thrush with counselling to avoid tongue biting. In addition, the patient reported dyspnoea described as a decrease in tolerance of his physical activities. Due to continued increase in tongue size and worsening dyspnoea, he underwent a tissue biopsy with findings consistent with amyloidosis. Further evaluation with a bone marrow biopsy revealed underlying multiple myeloma. Echocardiography revealed abnormal ventricular wall thickness, with a reduced left ventricular chamber size, dilated atria and Doppler findings with restrictive filling patterns indicative of cardiac amyloidosis. The patient was initiated on chemotherapy for his multiple myeloma and supportive therapy for his cardiac amyloidosis. Light-chain amyloidosis (AL) is a systemic disease characterised by irreversible deposition of amyloid in tissues throughout the body; when there is cardiac involvement, it can result in heart failure with a poor prognosis. Early diagnosis of cardiac amyloidosis can lead to prolonged survival.
- cardiovascular medicine
- medical education
- oncology
- ophthalmology
- otolaryngology / ENT
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Background
Cardiac amyloidosis is an aggressive and irreversible condition with a swift clinical course if not recognised promptly; however, diagnosis is often delayed or missed altogether. Amyloidosis and other infiltrative disorders can present with heart failure with preserved ejection fraction (HFpEF) phenotype. Given heart failure is the most common cause of hospitalisation in patients over 65 years of age, with an estimated 23 million people affected worldwide,1 half of that figure encompassing HFpEF,2 it stands that amyloidosis may be an underlying precipitant. One study performed autopsy evaluations of patients with an antemortem diagnosis of HFpEF and estimated that at least 5% of HFpEF represent undiagnosed cardiac amyloidosis.3 An understanding of the underlying pathophysiology is important in detecting clinical signs across affected organ systems as well as targeting treatment. Amyloidosis results from abnormal deposition of protein extracellular material from precursor proteins including light-chain (AL), wild-type transthyretin (ATTRwt), mutant transthyretin (ATTRm) and serum amyloid A (AA) derived proteins. Clinical examination findings such as macroglossia and periorbital bruising are common in AL cardiac amyloidosis and when it is associated with dyspnoea, it is pathognomonic for the diagnosis.4 5 It is essential to identify the precursor protein responsible for amyloid deposition because the therapy and clinical course differ among different cardiac amyloidoses. Early diagnosis is critical as it leads to improved outcomes if appropriate therapies are initiated early in the clinical course.6 There is a stark difference in clinical outcomes among patients with amyloid heart disease, with estimated 1 year survival of 38% for AL and up to 92% for ATTR amyolodisis.7 It is imperative that providers understand the clinical signs and symptoms of amyloidosis, which may present as disparate, seemingly unrelated findings. Here we discuss one such case of delayed diagnosis despite signs nearly pathognomonic for cardiac amyloidosis, and use it as a focus on which to offer providers a framework to recognise the symptoms across multiple organ systems in order to promote early diagnosis.
Case presentation
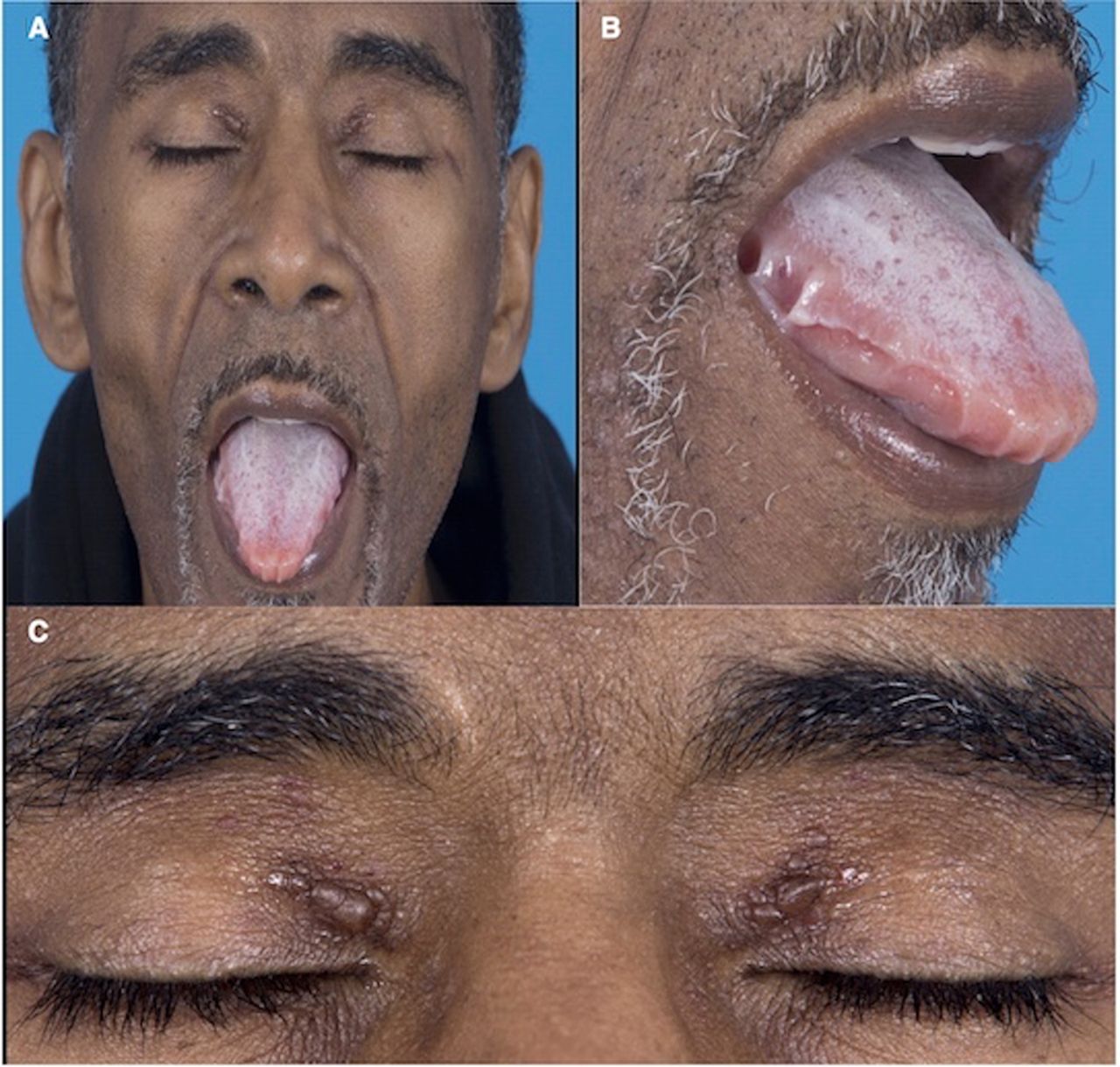
A 58-year-old man initially presented to his dentist with a chief complaint of tongue discomfort with unusual indentations (figure 1A,B). Due to his symptoms and findings he was referred to otolaryngology for further evaluation. Otolaryngology evaluation and examination was followed by treatment for oral thrush and tongue biting counselling, as it was determined that his indentations were likely due to unintentional biting. In addition to his tongue complaints, he reported a progressive dyspnoea with activity. The bilateral periorbital plaques (figure 1A,C) developed with the onset of his tongue discomfort. However, he did not seek medical attention for the periorbital plaques. His tongue symptoms remained stable for a year while the dyspnoea on exertion was not associated with paroxysmal nocturnal dyspnoea, orthopnoea or angina. He was re-evaluated by otolaryngology who did not notice overt macroglossia but biopsied the tongue given his persistent symptoms. Following his abnormal biopsy results, he was referred to haematology–oncology. During his initial evaluation with oncology, his clinical examination revealed findings of acute heart failure syndrome, characterised by a 6-month history of worsening dyspnoea on exertion and fatigue consistent with New York Heart Association class II symptoms with pitting lower extremity oedema, jugular venous distention while sitting upright, increased abdominal distention and mild bibasilar rales. The patient was hospitalised for acute management of his heart failure with cardiology consultation. He reported history of bilateral carpal tunnel syndrome status postsurgical release 2 years prior to the onset of his symptoms. He is a non-smoker and has no family history of cardiac infiltrative disease in first-degree relatives or first-degree relatives with a history of cardiomyopathies.

(A and B) Oral examination reveals characteristic scalloping at the lateral borders of the tongue with deep indentations adjacent to the premolars and molars. The patient experienced significant oral discomfort from the subsequent lingual pressure. (A and C) Periorbital ecchymosis, also called amyloid purpura, is evident on visual inspection. Vessel wall damage by amyloid deposition leads to fragility of the capillary beds and increased bleeding.
Investigations
Initial laboratory assay revealed a microcytic anaemia (haemoglobin 11.7 g/dL mean corpuscle value of 78.0fL), hyperlipidemia (an elevated LDL cholesterol of 148 mg/dL), hypoalbuminemia (3.3 g/dL), proteinuria (552 mg/dL), a mild elevation in serum troponin (0.050 ng/mL), an elevated proBNP (3407.0 pg/mL), normal hepatic enzymes, normal thyrotropin and a normal serum coagulation profile.
Histopathology from tongue biopsy demonstrated AL amyloid with positive Congo red stain and mass spectrometry. Immunofixation study revealed monoclonal free lambda light chain in gamma region. Free light-chain analysis was normal for kappa but markedly elevated for lambda at 2297 mg/dL. Bone marrow biopsy demonstrated 95% plasma cell myeloma with concomitant amyloidosis. Molecular cytogenetics indicated a variant FGFR3/IGH fusion resulting from the t(4:14) with deletion of one IGH gene region in 90.5% of the nuclei and monosomy 13q deletion in 93.5% of the nuclei. There was also minor tetraploid clone observed in 8.5% of the nuclei. The cytogenetic abnormalities noted are associated with an unfavourable prognosis in multiple myeloma (MM).
Transthoracic echocardiogram (figure 2A) revealed increased ventricular wall thickness with concentric hypertrophy, enlarged atria, small pericardial effusion and mild diffuse thickening of the mitral and tricuspid valves. Two-dimensional strain (figure 2B) using speckle-tracking echocardiography of the left ventricle demonstrated abnormal regional and average global longitudinal peak systolic strain rates with relative normal strain rates in the apex, consistent with apical sparing. Electrocardiogram (figure 3) was significant for low voltage in the limb leads, indeterminate axis, poor R wave progression in the precordial leads, left atrial enlargement and non-specific T wave changes.

(A) Transthoracic echocardiogram apical four chamber view reveals evidence of biatrial enlargement with ventricular hypertrophy, and mild diffuse thickening of the mitral and tricuspid valves. (B) Two-dimensional strain using speckle-tracking echocardiography of the left ventricle demonstrated abnormal regional and average global longitudinal peak systolic strain rates with relative normal strain rates in the apex, consistent with apical sparing.

{kind=link}
{kind=link}
{kind=link}
ECG with indeterminate axis, low limb lead voltage and poor precordial R-wave progression in cardiac AL (light-chain) amyloidosis.
Differential diagnosis
AL cardiac amyloidosis.
Multiple myeloma.
Hypertensive cardiomyopathy.
Fabry’s disease.
Treatment
The oncologist initiated therapy with bortezomib, lenalidomide and dexamethasone as part of his initial chemotherapy for his underlying MM. Cardiology directed therapy for heart failure primarily with supportive therapies with patient education for heart failure, diuretic therapy and initiation of doxycycline, for underlying AL cardiac amyloidosis. Further recommendations from cardiology included avoiding beta-blockers, ACE inhibitors to avoid decreases in cardiac output and avoid orthostatic hypotension. Further recommendation was provided for initiation of anticoagulation for any atrial arrhythmia and to avoid the use of digoxin.
Outcome and follow-up
One year later, patient is now status postinduction therapy (lenalidomide, bortezomib and dexamethasone), consolidation therapy with autologous haematopoietic stem cell transplantation, and on bortezomib maintenance therapy. His heart failure symptom resolved and he is no longer on diuretic. He is back to bowling regularly and will be travelling in July 2018 to Las Vegas for a bowling tournament.
Discussion
Light-chain (AL) cardiac amyloidosis is a rare disease with an annual incidence rate of 10.5 cases per million person-years.8 In AL cardiac amyloidosis, fibrillar proteinaceous material made by clonal plasma cell is deposited in the extracellular space of the myocardium. The abnormal clone of plasma cell overproduces lambda or kappa light chains (less commonly) in AL amyloidosis and they are highly resistant to degradation.5 9 10 Other precursor proteins of amyloid that could involve the heart in its disease course include ATTRwt, ATTRm and AA. Once cardiac amyloid is diagnosed, it is highly important to identify the precursor protein because the disease course, prognosis and therapy differ among different forms of amyloid.11 Amyloidosis is a systemic disease and early diagnosis is particularly important because once the patient develops heart failure, the median survival is 6 months.8 Early diagnosis and prompt initiation of current therapy could extend life and put the disease in extended remission.10
Diagnosing AL cardiac amyloidosis early is challenging because of its insidious course. Most patients are diagnosed when they present with overt symptoms of cardiac manifestation such as dyspnoea on exertion with associated peripheral oedema. However, there are some physical examination findings and clinical history that can help facilitate early diagnosis of AL cardiac amyloidosis. About 10% of patients with AL cardiac amyloidosis have macroglossia, which could present as apparent tongue enlargement or subtle tooth indentation on the lateral tongue.12 Macroglossia, in particular, is considered to be a hallmark of AL amyloidosis and is rarely observed in the other subtypes.12 13 This physical examination finding is present in our patient as reflected in figure 1A,B. Our patient did not present with overt macroglossia, which could have contributed to the limited differential on his initial presentation. However, the tooth indentation on his lateral tongue is evidence of tongue enlargement secondary to amyloid deposition. Periorbital bruising is also a common presentation in AL amyloidosis and when present with associated heart failure, it is pathognomonic for AL amyloidosis.5 The periorbital bruises can be elusive especially in dark skin individuals and the clinician has to examine the periorbital region and the eyelids closely for small bruises. Our patient did not display periorbital bruises but he had bilateral periorbital plaques in the superior medial canthus region. The plaques were not biopsied but the temporal presentation in his clinical course with the macroglossia is supportive of AL cardiac amyloidosis. Some patients could present with sensory neuropathy and surgical history for carpal tunnel syndrome that usually preceded onset of heart failure in AL cardiac amyloidosis by approximately 7 years.14 Our patient had a history of carpal tunnel syndrome and he was status postsurgical release of the median nerve by 2 years before onset of heart failure.
When amyloidosis is suspected, an ECG should be performed and the patient should be further evaluated with transthoracic echocardiogram. The ECG in AL cardiac amyloidosis usually reveals low voltage despite evidence of at times increase in myocardial thickening on echocardiography.15 The findings of low voltage on ECG usually precede heart failure symptoms in AL cardiac amyloidosis and may be present before ventricular wall thickness is noted on echocardiogram.16 Although there is a heavier burden of myocardium amyloid deposition in ATTR than in AL cardiac amyloidosis, low voltage QRS complexes are less common in ATTR.7 Echocardiogram typically reveals ventricular wall thickening and mildly thickening valves. The left ventricular wall thickness frequently surpass 15 mm and a wall thickness of greater than 18 mm can be seen in AL cardiac amyloidosis although more commonly in ATTR.11 The incongruity of low voltage ECG with increased wall thickness on echocardiogram should raise suspicion for cardiac amyloidosis.7 16
AL amyloidosis is a systemic disease and evidence of extracardiac involvement could manifest on routine basic laboratory assay. In 2004, the 10th International Symposium on Amyloid and Amyloidosis was held and guidelines were developed to define organ involvement, thus establishing uniform reporting criteria. Our patient demonstrated evidence of renal involvement with (1) biopsy proof of amyloid at an alternate site (tongue) with (2) laboratory evidence of renal dysfunction to include proteinuria greater than or equal to 0.5 g per day. Cardiac involvement was established by meeting the criteria of echo wall thickness greater than 12 mm with no other identifiable cause. Soft tissue involvement was established with (1) tongue enlargement with subsequent biopsy and (2) a history of carpal tunnel syndrome.17 Other laboratory markers support these findings while not being strict guideline criteria. Patients with renal involvement can present with nephrotic syndrome evident by hypoalbuminaemia, hypercholesterolemia and proteinuria. Patients with cardiac amyloidosis usually have disproportionately elevated NT-proBNP with a chronic mildly elevated troponin.6 Immunofixation study and serum-free light-chain assay should be measured in patients suspected of having amyloidosis. A normal ratio of free light-chain kappa/lambda makes AL amyloidosis less likely given that over 90% of untreated AL amyloidosis have abnormal free light-chain kappa/lambda ratio.5 A prognostic staging system was created in 2004 using troponins and NT-proBNP and was revised in 2012 to incorporate free light-chain ratios. Known as the Mayo staging system, patients with AL amyloidosis are assigned stage 1, 2, 3 or 4 based on the presence of 0, 1, 2 or 3 prognostic signs (cardiac troponin T≥0.025 ng/mL, NT-proBNP ≥1800 pg/mL and free light-chain difference ≥18 mg/dL) with median survivals of 94.1, 40.3, 14 and 5.8 months, respectively. In this case, our patient had elevated cardiac troponins and NT-proBNP with an abnormal kappa/lambda ratio consistent with Mayo stage IV disease.6 18
Bone marrow biopsy is critical in suspected amyloidosis to rule out associated MM. Historically, these two diseases have been categorised as being on a spectrum of plasma cell dyscrasia with considerable overlap; amyloid fibrils in AL amyloidosis are derived from immunoglobulin light chains produced from a plasma cell clonal process such as that seen in MM.19 The hallmark of the amyloid monoclonal light chain, in comparison with MM, is its propensity to form insoluble fibrils with specific tropism for various organs rather than primarily infiltrating bone marrow. Hence, the diagnosis of AL amyloidosis requires histological confirmation of organ tissue with a biopsy specimen staining positive with Congo red and demonstrating apple green birefringence under polarised light. However, it is estimated that 10% of patients with MM may have AL amyloidosis and a similar percentage of those with AL amyloidosis may have MM. Therefore, AL amyloidosis may share numerical chromosomal changes with MM, as is evident in our patient who was found to be positive for t(4;14) a finding associated with MM. Guidelines advise that a distinction between MM and AL lies in the percentage of plasma cell involvement, with an estimated 18% of patients with AL amyloidosis having >20% plasma cells in the bone marrow, and the majority (60%) having <10% involvement.19 20 Thus, bone marrow of AL amyloidosis patients usually has less than 20% of plasma cells in comparison to MM.21 Abdominal fat pad biopsy or gum tissue biopsy with appropriate staining allows for the determination of amyloid; these areas are typically biopsied first because they are easier to access and therefore less invasive. However, if amyloid is not visualised on Congo red staining and clinical suspicion remains high, other organs may subsequently be biopsied using invasive methods. In our patient, clinical suspicion of tongue involvement was grossly evident so the initial biopsy was of lingual tissue. Knowledge of additional organ involvement may also be desired to direct treatment.20 The tissue should be processed and stained in an experienced centre to avoid false negatives and false positives that could occur in less experienced centres.21–23
The primary therapy of AL cardiac amyloidosis consists of chemotherapy directed at the underlying plasma cell dyscrasia and supportive heart failure management. Bortezomib, a proteasome-inhibiting agent is usually combined with dexamethasone in patients with light-chain cardiac amyloidosis and well tolerated.24 25 An immunomodulatory agent, lenalidomide can be used in AL cardiac amyloidosis and patient usually have paradoxical elevation of proBNP even in the setting of haematologic improvement and the mechanism is currently unknown.18 26 It is important to involve a cardiologist in the management of the heart failure because several parts of the management differ from routine conventional heart failure management. Diuretics are used primarily for managing heart failure symptoms in AL cardiac amyloidosis. Most patients with AL cardiac amyloidosis usually have asymptomatic low systolic blood pressures and should not be given fluids. The low blood pressure is secondary to reduced cardiac output and decreased peripheral tone.5 Beta-blockers, ACE inhibitors and angiotensin-receptor blockers may aggravate hypotension and are usually not well tolerated. There is no role for digoxin in AL cardiac amyloidosis patients with heart failure and digoxin toxicity has been observed with therapeutic digoxin levels in AL cardiac amyloidosis patients.27 In addition to possible concomitant nephrotic syndrome, patient with AL cardiac amyloidosis are prone to atrial thromboembolism, even without evidence of any atrial arrhythmia; thus, there is a potential indication for systemic anticoagulation to prevent cardioembolic events. Patients with AL cardiac amyloidosis who are in sinus rhythm but have evidence of left atrial mechanical dysfunction on echocardiogram are candidates for systemic anticoagulation.5Patients with AL cardiac amyloidosis rarely have non-sustained ventricular arrhythmias and sudden cardiac death if it occurs usually secondary to intense bradycardia or pulseless electrical activity.28 There is no role for prophylactic implantable cardiac defibrillator in AL cardiac amyloidosis patients because there is diminutive evidence that long-term survival is affected.29 30
Prompt recognition and diagnosis of cardiac amyloidosis is imperative and still elusive; if untreated, the median survival of patients with cardiac AL amyloidosis is 6 months from the onset of heart failure.24 Given the swift mortality if unrecognised, cardiac amyloidosis should be in the differential diagnosis in patients with new onset of HFpEF in the absence of risk factors for heart failure.
Learning points
Any patient with overt macroglossia or lateral tongue indentation with or without associated periorbital bruise or plaque and symptoms of dyspnoea should be evaluated for AL cardiac amyloidosis.
Cardiac amyloidosis with overt heart failure symptoms carries poor prognosis but the earlier it is diagnosed and treated, the better their prognosis.
Identifying the precursor amyloid protein in amyloidosis is important because disease course and therapy differ among different forms of amyloid.
Discrepancy observed on low voltage ECG with associated ventricular wall thickness on echocardiogramis highly suspicious for cardiac amyloidosis.
Prophylactic cardiac defibrillator is not indicated in AL cardiac amyloidosis.
Acknowledgments
Medical Photo, San Antonio Military Medical Center, San Antonio, Texas.
References
Footnotes
Patient consent for publication Obtained.
Contributors MUW involved in conception, logistics/planning (obtained informed consent, image acquisition with medical images), drafted initial manuscript, revision. CEM participated in writing the manuscript, revision. RSG drafted manuscript revision, approval of final version. EF involved in planning, manuscript revision, recommended BMJ Case Report, planning and approval of final version.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.