Article Text

Statistics from Altmetric.com
Case description
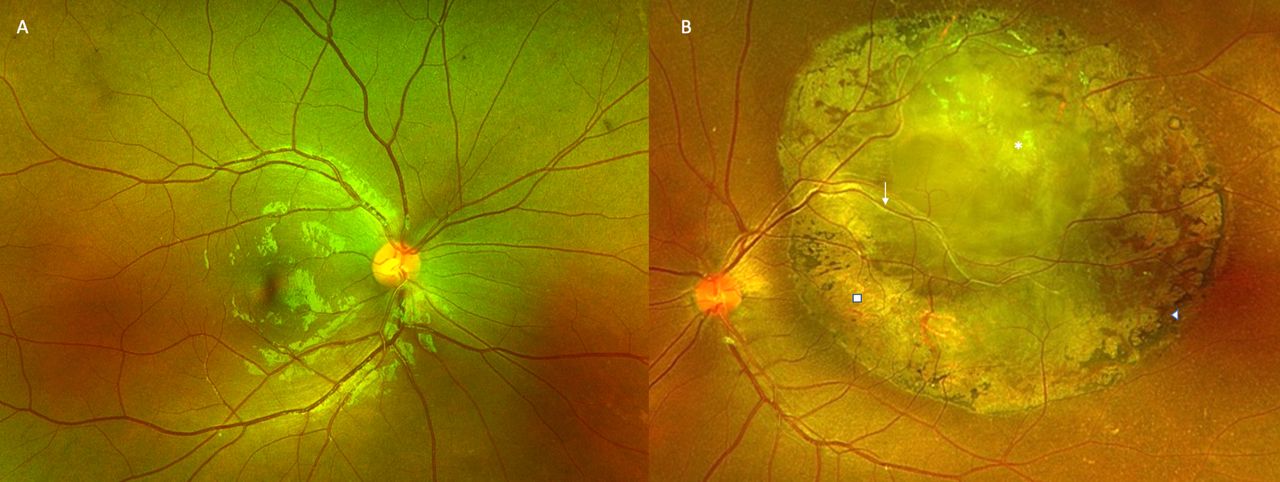
A young woman in her early 20s, with unremarkable past ophthalmic and family history, presented to the outpatient services with diminution of vision OS since childhood. On clinical exam, the best-corrected visual acuity was 20/20 OD and 20/200 OS with normal intraocular pressures. Gross examination revealed 30° exotropia OS with unremarkable anterior segment examination on slit-lamp biomicroscopy. Fundus examination revealed normal fundus OD (figure 1A) with a well-defined, white-grey, translucent retinal mass of the posterior pole with feeder vessels, perilesional pigmentary changes and chorioretinal atrophy OS (figure 1B). Autofluorescence imaging revealed normal retinal autofluorescence OD (figure 2A) and a large central area of hypoautofluorescence with a central island of hyperautofluorescence OS (figure 2B). The paternal fundus examination revealed vascular sclerosis and avascularity in the temporal peripheral retina with pigment clump (non-pertinent to the index case) and normal posterior poles OU (figure 3A,B). The maternal fundus examination was unremarkable OU (figure 3C,D). Ocular coherence tomography (OCT) (figure 4) suggested a superficial retinal mass lesion with homogeneous internal reflectivity and occasional intralesional hyporeflective spaces with posterior hyper-reflectivity OS. Ultrasonography (figure 5) revealed a corroborating retinal mass lesion of 6.3 mm × 1.8 mm in greatest dimensions with hyperechoic spikes corresponding to intralesional calcification. A clinical diagnosis of retinocytoma was made. The prognosis was explained and the patient continues to be under routine follow-up.

Ultra widefield fundus photograph depicting normal fundus OD (A) and grey-white, fleshy translucent retinal mass (asterisk) with pigment epithelial alteration (arrow head), chorioretinal atrophy (block) and vessels feeding into the lesion (line arrow) (B).

Autofluorescence imaging showing normal retinal autofluorescence OD (A) and a large central area of hypoautofluorescence with a central island of hyperautofluorescence OS (B).
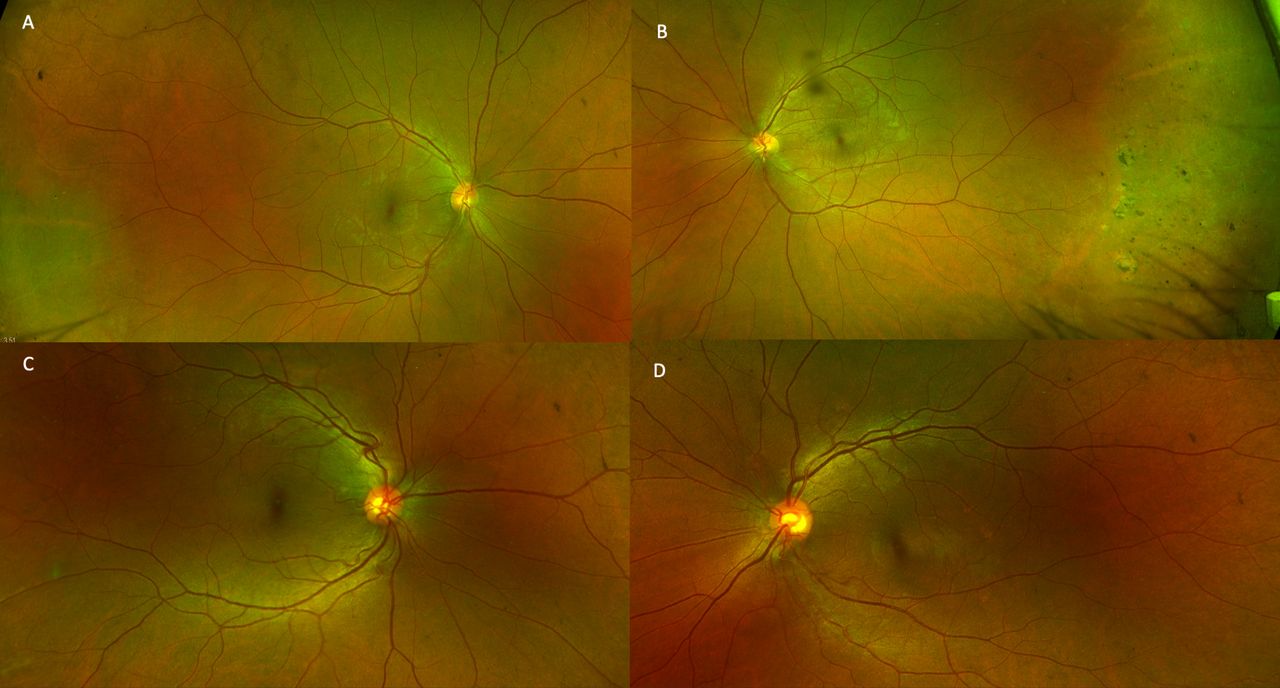
Fundus examination of the father depicting vascular sclerosis and avascularity in the temporal peripheral retina OU (A, B). The maternal fundus revealed an unremarkable posterior pole (C, D).
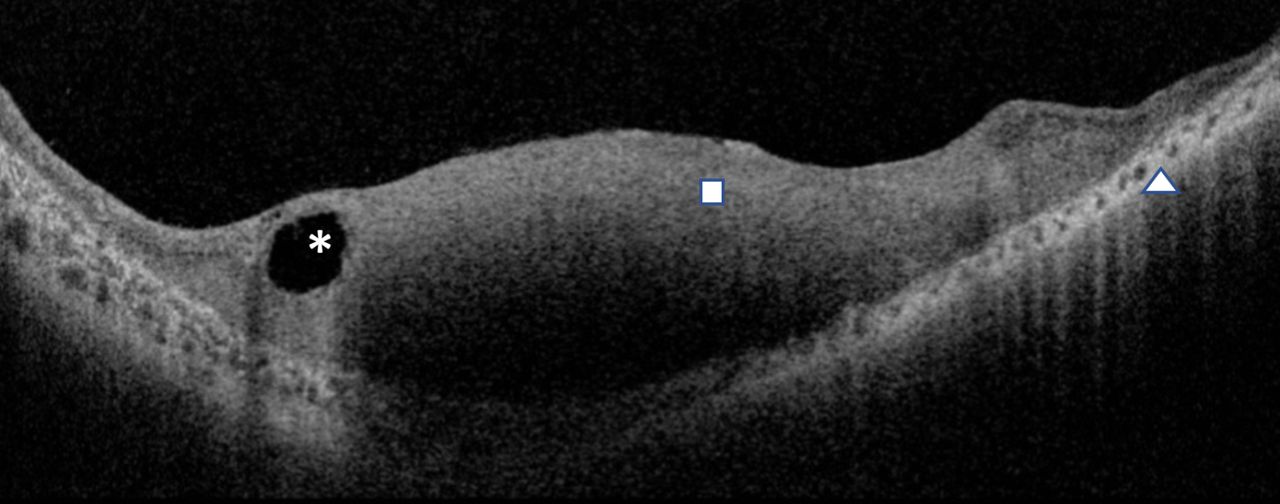
Optical coherence tomography depicting homogeneous retinal mass lesion (block) with intralesional hyporeflective spaces overlying areas of posterior hyper-reflectivity (asterisk) and chorioretinal atrophic changes (arrow head).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Ultrasound scan showing hyperechoic spikes corresponding to intralesional calcification.
The differential diagnoses to be considered in cases of retinocytoma include retinal astrocytic hamartomas and retinoblastomas. A meticulous clinical examination supplemented with investigations can facilitate differentiation of retinal hamartomas from retinocytomas. A multilobulated dome-shaped elevated/semitranslucent flat lesions with lack of pigment epithelial alteration and optically empty spaces on OCT appearance suggests a diagnosis of astrocytic hamartoma.1 Unfortunately, the only definitive way of differentiating retinocytomas from retinoblastomas are subsequent follow ups for any progressive lesional changes and investigations play little, if any, effect to the same.2
In their descriptions, Gallie et al described retinocytomas as grey, translucent lesions of the posterior pole with intralesional calcification, pigment epithelial alteration, and areas of chorioretinal atrophy harbouring mutations in both the alleles of the RB1 gene (Rb-/-).3 4 The mutated protein, pRb, disrupts the normal checkpoint inhibition of the cell cycle affecting unchecked cell proliferation.5 Despite the common RB-/- genotype as shared with retinoblastomas, retinocytomas are benign, well-differentiated mass lesions sharing common origins in the retinocyte precursors.1 6 The benign nature of these lesions was earlier attributed to a late-stage hit of the second allele, varying retention of wild-type protein function, and associated low penetrance.3 7–9 Recent genetic analysis of patients with retinocytoma, however, suggests mutated RB1 gene-induced genomic field instability with high expression of senescence-associated proteins as the mutations underpinning the benign attribute of retinocytoma. Accruing further mutations that induce oncogenic instability and/or reduced expression of senescence-associated proteins may portend the malignant transformation of these lesions.8 These malignant changes are not uncommon and occur in a reported 15% of the patients.10 Because of their treatment-resistant-well differentiated attribute, as opposed to retinoblastomas, careful observation of the lesion may obviate unwarranted surgical/medical interventions.11–13 Routine follow-up, therefore, becomes imperative in affecting early interventions and better treatment outcomes at the advent of such malignant transformations.
Learning points
Retinocytomas are benign, well-differentiated tumours originating from the retinocyte precursors.
Despite their benign attribute, they have the potential for malignant transformation to retinoblastoma.
Ethics statements
Patient consent for publication
Footnotes
Contributors MS collected the data and prepared the draft. PJ collected the data. RC reviewed conceptualised the idea, proof read the manuscript and provided valuable suggestions. NS reviewed the manuscript.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Case reports provide a valuable learning resource for the scientific community and can indicate areas of interest for future research. They should not be used in isolation to guide treatment choices or public health policy.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.