Article Text

Abstract
Osteogenesis imperfecta is a congenital disease that presents with varying degrees of connective tissue symptoms, including susceptibility to fracture, growth disorders and hearing loss. Here, we discuss a case in which macular neovascularisation (MNV) resulted in metamorphopsia and decreased visual acuity in a patient with osteogenesis imperfecta exhibiting a novel COL1A1 gene mutation (p.Tyr165*). The patient was a woman in her 30s who reported experiencing distorted vision and diminished visual acuity in her right eye for 1 month as well as a history of hearing loss. Rapid improvements in exudative changes and suppression of relapse were achieved after only two intravitreal injections of ranibizumab. Furthermore, since MNV occurred slightly inferior to the fovea centralis, improvements in visual acuity were better than previously reported. As fragility of Bruch’s membrane represents the basis of onset, recurrence and relapse are likely in patients exhibiting MNV, highlighting the need for regular follow-up.
- Retina
- Orthopaedics
- Macula
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
Osteogenesis imperfecta is a congenital disease that presents with varying degrees of connective tissue symptoms as well as susceptibility to fractures and progressive bone deformities due to systemic bone fragility, with an incidence ranging from approximately 1 in 20 000 to 1 in 30 000. The 2019 edition of the Nosology & Classification of Genetic Skeletal Disorders classifies osteogenesis imperfecta into type 1 (non-deformation with persistently blue sclerae), type 2 (perinatal lethal form), type 3 (progressively deforming type), type 4 (moderate form),1 type 5—which is accompanied by interosseous calcification/hyperplastic callus—and other types. Type 1 is the most common type of osteogenesis imperfecta among adults.
In more than 90% of cases, osteogenesis imperfecta is caused by genetic mutations (COL1A1, COL1A2) affecting type I collagen, which is a major component of connective tissue, especially in the bones and ligaments. Clinically, patients with osteogenesis imperfecta exhibit susceptibility to fracture, growth disorders, hearing loss, hyperextension of the joints and skin and valvular disease. In 1965, Manschot demonstrated that osteogenesis imperfecta is associated with thinning of the cornea and sclera as well as blue scleral findings.2 In 2014, Wallace et al3 reported an association between osteogenesis imperfecta and open-angle glaucoma. Other typical ocular findings are myopia and keratoconus. However, only three case reports have described macular neovascularisation (MNV) occurring in the context of osteogenesis imperfecta.4–6
In this report, we discuss a case in which MNV resulted in metamorphopsia and decreased visual acuity in a patient with osteogenesis imperfecta, exhibiting a novel COL1A1 gene mutation (p.Tyr165*).
Case presentation
A woman in her 30s who reported experiencing metamorphopsia and diminished visual acuity in her right eye for 1 month was referred to Oita University Hospital for detailed examination. Although she had four fractures during her childhood, she had no history of systemic illness. She also disclosed that her otolaryngologist had diagnosed hearing loss, the cause of which remained unknown. Blue sclerae were also observed in two of the patient’s children (both male). The elder son, who was 9 years old, had several histories of fractures and was short in stature. The younger son, who was 7 years old, had a history of fractures in early childhood.
Investigations
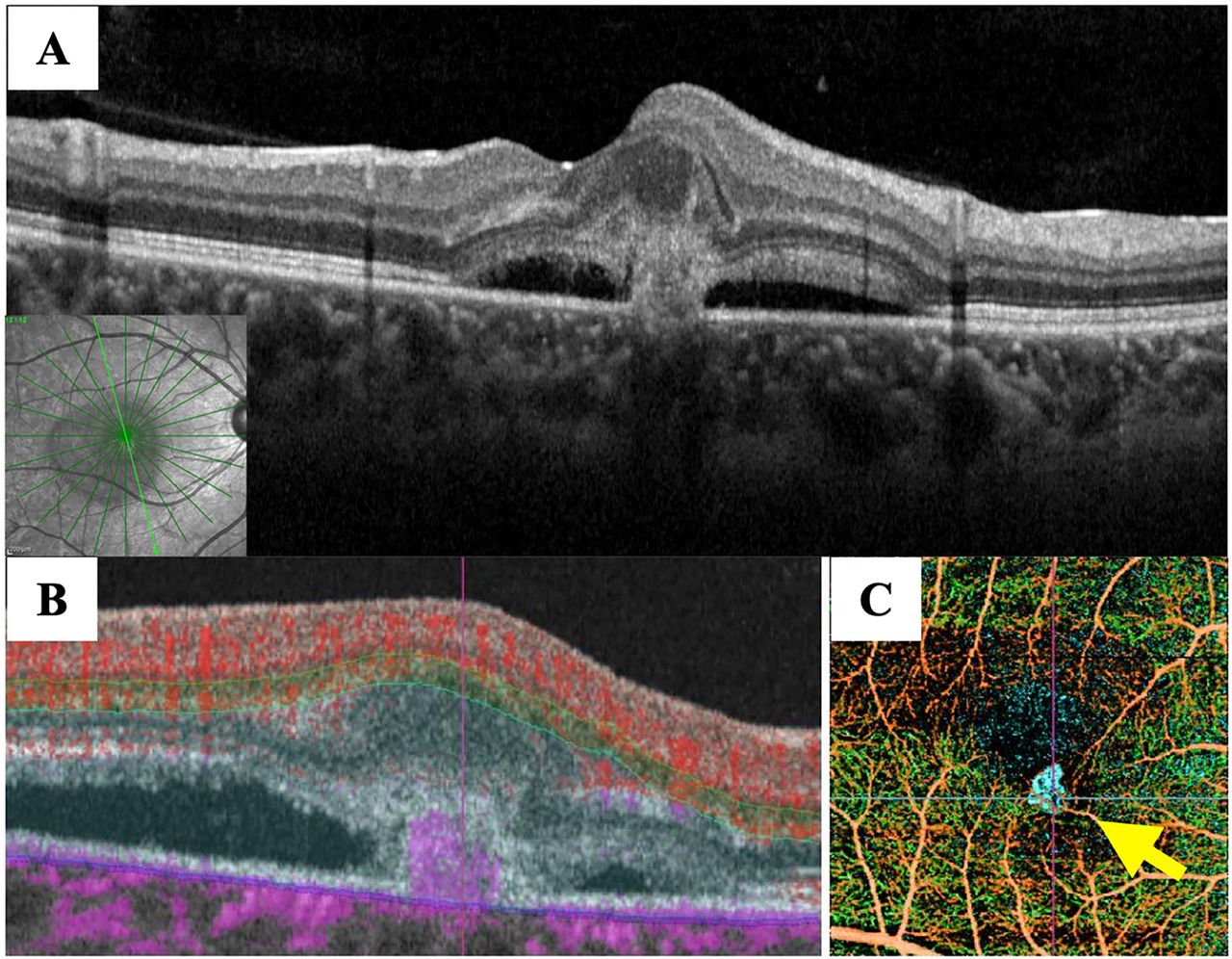
Decimal visual acuity was 0.4×−3.5D in her right eye and 1.2×−3.5D in her left eye. Ocular pressure was 14 mmHg in her right eye and 15 mmHg in her left eye. Axial length was 23.97 mm in her right eye and 23.96 mm in her left eye. Both eyes exhibited blue sclerae (figure 1). Anterior segment optical coherence tomography (OCT) indicated that corneal thickness was 421 µm in the right eye and 427 µm in the left eye. There were no noteworthy findings in the optic media, and her left fundus was normal. However, her right fundus exhibited a greyish-white exudative lesion with haemorrhage, extending into the parafovea and serous retinal detachment (size: 2 papillae) (figure 2A). Fundus autofluorescence images showed a faint hypofluorescence consistent with the range of serous retinal detachment and a hypofluorescence in the area of the subretinal haemorrhage (figure 2B). A highly reflexive exudative neovascular complex above the retinal pigment epithelium was also observed on OCT images (figure 3A). There was no local thickening of the choroid or compression of the choriocapillaris, and the thickness of the choroid was almost normal. OCT angiography B-scan clearly demonstrated a blood flow signal consistent with the exudative lesion in the parafovea (figure 3B). Reconstructed composite images showed clear subretinal vascular networks (figure 3C). Based on the above, the patient was diagnosed with type 2 MNV.

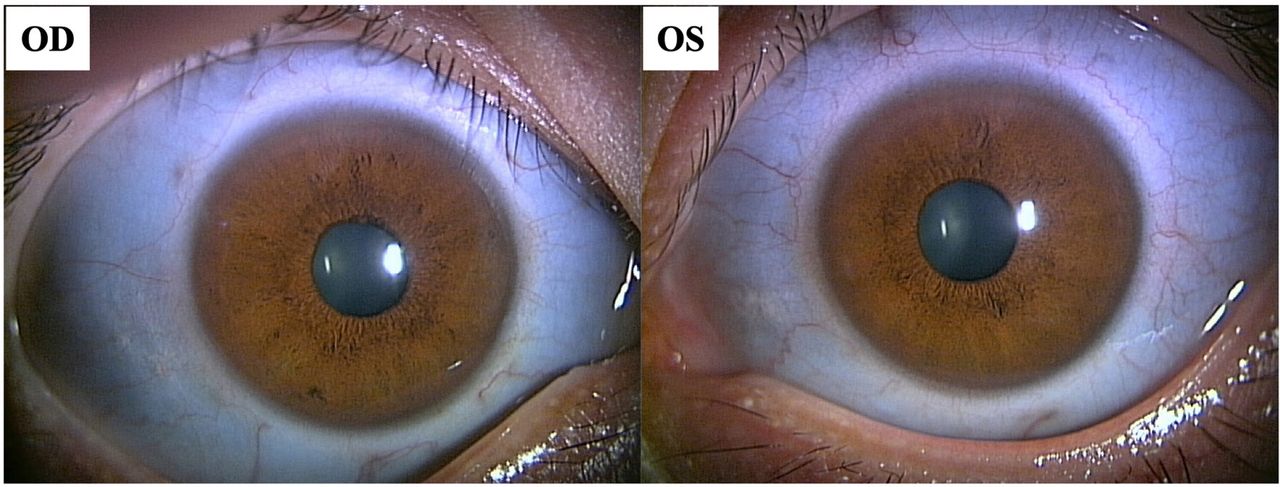
Photograph of the anterior segment of the eye at the first visit (blue sclerae). The sclera was blue in both eyes.

Findings in the right fundus at the first visit. (A) Photograph of the fundus at the first visit. Serous retinal detachment, subretinal haemorrhage and greyish-white exudative lesions were observed in the parafoveal lesion. Serous retinal detachment was observed in the macula. (B) Fundus autofluorescence images. A faint hypofluorescence signal was observed consistent with serous retinal detachment. The site of subretinal haemorrhage also exhibited hypofluorescence.

Optical coherence tomography (OCT) and OCT angiography (OCTA) images obtained at the first visit. (A) OCT. Highly reflexive exudative lesions were observed to extend beyond the retinal pigment epithelium. (B) OCTA B-scan clearly demonstrated a blood flow signal consistent with the exudative lesion in the parafovea. (C) The composite image of OCTA shows the clear subretinal vascular networks (yellow arrow).
Differential diagnosis
Osteogenesis imperfecta was suspected based on the presence of blue sclerae, the patient’s history of hearing loss/fracture and the results of a bone mineral density test. Relative to the Young Adult Mean, bone mineral densities for the right femur, left femur and lumbar spine were 62%, 60% and 72%, respectively, which were lower than the standard value of 80% or more. Genetic analysis via next-generation sequencing revealed a novel variant of a nonsense mutation in COL1A1 (heterozygous, c.495T>A, p.Tyr165*). Furthermore, the relevant spot was confirmed by Sanger method (figure 4). Based on the above results, a definitive diagnosis of osteogenesis imperfecta was made.

We performed Sanger sequencing of the coding exon of the COL1A1 gene using following primers: 5′-TGATCTGGACCTCCCAAGC-3′(sense) and 5′-CACATCACACCAGGAAGTGC-3′(antisense). COL1A1 mutation (heterozygous, c.495T>A, p.Tyr165*) was confirmed (red arrow).
Treatment
After ruling out pregnancy, an intravitreal injection of ranibizumab was administered on the day of the visit.
Outcome and follow-up
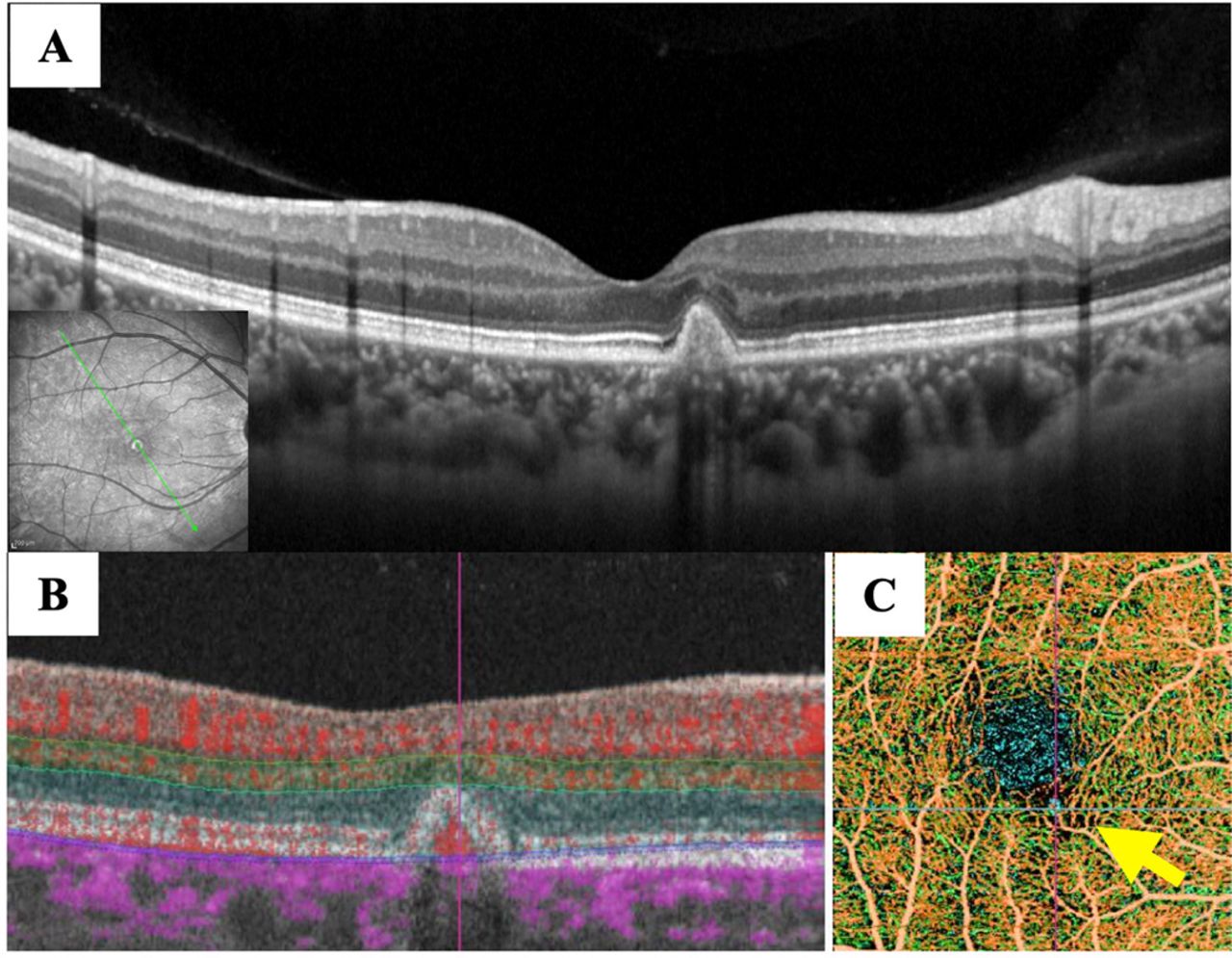
One month after the initial diagnosis, serous retinal detachment had disappeared, and corrected visual acuity had improved to 0.7 in the right eye. Two months later, corrected visual acuity in the right eye had improved to 1.2. OCT and OCT angiography revealed a marked reduction in the neovascularisation signal when compared with that observed before treatment (figure 5). However, 1 month later, mild serous retinal detachment was observed again, although corrected visual acuity in her right eye remained unchanged at 1.2. A second injection of ranibizumab into the vitreous body promptly attenuated the exudative changes. After 25 months, she has had no relapse of the lesion, and corrected visual acuity in the right eye remains unchanged at 1.2.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Optical coherence tomography (OCT) and angiography (OCTA) images obtained 2 months after treatment. (A) OCT. The exudative changes had subsided and macular neovascularisation (MNV) was covered with the retinal pigment epithelium. (B) OCTA B-scan clearly demonstrated a blood flow signal consistent with MNV reduction. (C) The composite image of OCTA shows that MNV had diminished dramatically (yellow arrow).
Discussion
The present report is the first to discuss a case of MNV associated with osteogenesis imperfecta in Japan. Patients with osteogenesis imperfecta exhibit blue sclerae due to scleral thinning and transparency of the choroid, resulting from collagen abnormalities. This sign can also be observed in patients with Ehlers–Danlos syndrome. In general, in patients with osteogenesis imperfecta, abnormalities in type I collagen reduce ocular rigidity, resulting in axial elongation of the globe and myopia associated with the development of posterior staphyloma.
Bruch’s membrane is composed of five layers. From the retinal side, these layers are the basement membrane of retinal pigment epithelial cells, the inner collagen layer, the elastic plate, the outer collagen layer and the basement membrane of choroidal capillaries. Since the inner and outer collagen layers contain type I collagen, COL1A1 mutation may explain weakening of Bruch’s membrane in patients with osteogenesis imperfecta. In all previous reports of MNV associated with osteogenesis imperfecta, the presence of lacquer cracks was confirmed.4–6 Rishi et al reported that a 12-year-old girl developed a lacquer crack after intravitreal injection of bevacizumab, speculating that the temporary increase in intraocular pressure after intravitreal injection or contraction of the retinal pigment epithelium associated with the retraction of MNV caused the Bruch’s membrane to rupture. Bellanca et al5 reported lacquer cracks in a 28-year-old man with osteogenesis imperfecta who did not use antivascular endothelial growth factor drugs. Despite treatment for MNV, the patient developed lacquer cracks, which may have been due to decreased tension and ankylosis of the eyeball resulting from eyeball elongation.
In the present case, no clear lacquer cracks were observed either before or after treatment. Although refractive errors can develop over a long period of time, the error observed in our patient was smaller than that indicated in previous reports, and no conspicuous extension of the ocular axis was observed. In addition, we speculate that the nonsense mutation (p.Tyr165*) in COL1A1 detected in our patient was not a dominant-negative mutation but instead represented a null allele due to nonsense codon-mediated mRNA degradation, resulting in haploinsufficiency. Therefore, although susceptibility to fracture and mild hearing loss were observed in the present case, systemic symptoms remained mild and did not interfere with daily life. We believe that lacquer cracks did not occur given that the patient exhibited only mild weakening of Bruch’s membrane.
Some reports have described genetic mutations affecting other types of collagen, which have also been associated with fragility of Bruch’s membrane. Corominas et al7 reported an association between exudative age-related macular degeneration (AMD) and COL8A1 gene mutation. According to their report, hyperpigmentation of the macula occurred in patients with early AMD exhibiting the COL8A1 mutation, who exhibited a greater degree of myopia than those without the mutation. Previous mouse studies have suggested that structural changes in Bruch’s membrane associated with the COL8A1 mutation lead to the onset of AMD. Thus, abnormalities in collagen fibres may lead to fragility of Bruch’s membrane, thereby resulting in structural changes and contributing to MNV. Indeed, we speculate that such weakening occurred in our patient due to changes in type I collagen in both the inner and outer layers, resulting from COL1A1 mutation.
Fortunately, in the present case, rapid improvement of the exudative changes and suppression of relapse were achieved with only two intravitreal injections of ranibizumab. Furthermore, since MNV was located slightly inferior to the fovea centralis, improvements in visual acuity were better than those reported for previous cases.4–6 Since fragility of Bruch’s membrane represents the basis of onset, recurrence and relapse are likely in patients exhibited MNV, highlighting the need for regular follow-up.
In conclusion, we discussed a case of MNV associated with osteogenesis imperfecta due to a nonsense mutation in COL1A1 (p.Tyr165 *). We believe that thinning of the cornea and sclera decreased the rigidity in the globe, and weakening of Bruch’s membrane due to abnormalities in type I collagen contributed to the onset of MNV in our patient.
Patient’s perspective
(The author translated the patient’s perspective into English.)
From an early age, I felt that my eyes were of a different colour from the people around me. I did not really care because I thought this was because the pigment of my body was lighter than the others.
Since I was in elementary school, I had repeated fractures and my wrists and fingers remained bent. My vision gradually became myopic. I had never been told that these are symptoms of this disease (osteogenesis imperfecta).
This time, my field of vision suddenly became unclear, landscapes were distorted, and my vision became blurry; it became difficult for me to see. I even hit the wall several times. I went to an ophthalmologist because I was afraid that I would lose my eyesight.
For the first time, I learnt the blue colour of the white of my eye (conjunctiva), the repeated fractures I experienced and macular neovascularisation in my eyes were symptoms of osteogenesis imperfecta.
The two injections I received into my eyes were very scary. Thanks to that I am getting better and living the same life as before. However, I am worried that it may recur every time I come to the hospital for a regular check-up.
I was worried that my two sons had inherited it, so my sons also underwent genetic testing. Turns out that both of them had inherited the disease; this shook me more than when I discovered that I had the disease.
I really did not want them to inherit it. It took me some time to make up my mind that I had to deal with this illness along with my two children.
Since both my children have low bone density, they experience several fractures and sprains. Hence, I have spoken with the school teachers and have told them that my children need to avoid strenuous exercises and that they should not bump into other people. I think my children will be more anxious because they are planning to receive intravenous drip treatment, but I would like to face this illness and live.
Learning points
Osteogenesis imperfecta was suspected based on the presence of blue sclerae, the patient’s history of hearing loss, fracture and the results of a bone mineral density test.
Rapid improvement of the exudative changes and suppression of relapse were achieved with only two intravitreal injections of ranibizumab.
Thinning of the cornea and sclera decreased the rigidity in the globe, and weakening of Bruch’s membrane due to abnormalities in type I collagen may have contributed to the onset of macular neovascularisation in our patient.
Since fragility of the Bruch’s membrane represents the basis of onset, recurrence and relapse are likely in patients exhibited macular neovascularisation, highlighting the need for regular follow-up.
Ethics statements
Patient consent for publication
Acknowledgments
We would like to thank Editage (www.editage.com) for English language editing.
Footnotes
Contributors YS, KK and YT worked up and diagnosed the patient. YS and KK performed imaging, wrote the manuscript, and YT and TK critically revised it. YS and KK are the guarantors of the paper.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Case reports provide a valuable learning resource for the scientific community and can indicate areas of interest for future research. They should not be used in isolation to guide treatment choices or public health policy.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.