Article Text

Abstract
A man in his 50s with resistant hypertension and history of Langerhans cell histiocytosis (LCH) was referred to rheumatology after suspicion of inflammatory arteritis was raised. This followed detection of bilateral renal artery stenosis during investigation for severe hypertension refractory to medical therapy. CT angiography revealed diffuse wall thickening of the abdominal aorta, in keeping with an aortitis. However, there was no serological or clinical evidence suggestive of a vasculitic process. Medical history included cranial diabetes insipidus, subclavian artery stenosis and spinal stenosis requiring surgery, over the course of 8 years. These findings led to consideration of Erdheim-Chester disease (ECD), a form of non-Langerhans cell histiocytosis, where there is abnormal proliferation of histiocytes which causes tissue fibrosis and sclerosis of the long bones. Subsequent plain radiographs of the long bones revealed appearances consistent with a diagnosis of ECD. Thus, a diagnosis of an LCH/ECD overlap syndrome was made.
- hypertension
- interventional radiology
- renal medicine
- rheumatology
- vasculitis
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
Erdheim-Chester disease (ECD) is an extremely rare condition and a form of non-Langerhans cell histiocytosis (LCH).1 It is a multisystem disorder characterised by an accumulation and infiltration of histiocytes in tissue. ECD belongs to a group of conditions known as rare histiocytoses and has been recently recognised as a neoplastic disease. The hallmark of ECD is symmetric osteosclerosis of the long bones due to the build up of CD68+ and CD1a− histiocytes, which manifests as bone pain, with leg pain being the most common symptom.2 3 ECD is a heterogeneous disease, a great mimicker and is underdiagnosed.1 As ECD is multisystemic, vascular involvement may mimic large vessel vasculitis (eg, Takayasu’s arteritis). We report the case of a male patient investigated for large vessel vasculitis, who was eventually diagnosed with an ECD and LCH overlap syndrome, 8 years after initial presentation. This case was particularly thought-provoking due to the challenge and complexity of the diagnosis and the multiple and varied complications.
Case presentation
This report describes a male lawyer in his 50s, presenting with a history of hypertension, cranial diabetes insipidus (DI), subclavian stenosis, spinal stenosis and a diagnosis of Langerhans cell histiocytosis (LCH).
Medications included bisoprolol 10 mg daily, amlodipine 5 mg daily, doxazosin 8 mg daily, simvastatin 20 mg nocte and minoxidil 400 mg daily. He was a non-smoker, who drank less than 20 units of alcohol per week. Family history was unremarkable.
The initial diagnosis of cutaneous LCH was made following presentation with alopecia and patchy erythema affecting the head, neck and torso. Skin biopsy findings were consistent with LCH and he was treated with immunosuppression, using azathioprine, which was continued for several years. That same year he was diagnosed with central diabetes insipidus (DI) and prescribed desmopressin. The DI was complicated by intermittent severe hyponatraemia, which on one occasion induced an epileptiform seizure. In the following year, he developed back pain, which was related to dural thickening and a tumour involving the cranial dura and spine, which required spinal decompression surgery. Biopsy of the spinal tumour demonstrated inflammatory changes only. Cladribine chemotherapy was commenced, after the spinal cord histiocytic process was operated on.
Seven years after the initial diagnosis of cutaneous LCH, the patient was noted to have developed refractory hypertension. The refractory hypertension was investigated with CT imaging which revealed changes suggestive of aortitis, with arterial wall thickening and bilateral renal artery stenosis, leading to a rheumatology referral. On review, the patient did not have weight loss, fevers or night sweats. There were no symptoms of claudication, but mobility had declined since the operations for spinal stenosis. He did not have bony pain, chest pain or dyspnoea.
On examination, blood pressure (BP) was raised in both arms. The left arm BP measured 140/100 mm Hg and the right arm BP was 180/110. The patient remained hypertensive despite medical management with: bisoprolol 10 mg daily, amlodipine 5 mg daily, doxazosin 8 mg daily and minoxidil 400 mg daily. ACE inhibitors were not used in light of the severe renal artery stenosis, as they may precipitate deteriorating renal function. The subclavian pulse on the left was weaker than the right, in keeping with previous subclavian stenosis. Pedal pulses were palpable. The peripheries were warm and well perfused with no trophic changes. There was no detectable lymphadenopathy. The remainder of the examination was unremarkable.
Investigations
Review of the CT angiography revealed diffuse wall thickening of the abdominal aorta (figure 1A), a left subclavian artery stenosis, severe stenosis of a single right renal artery and severe ostial stenosis of the superior left renal artery (figure 1B), with complete occlusion of the inferior left anomalous branch, with stenoses also affecting the coeliac axis and inferior mesenteric artery. Laboratory investigations demonstrated normal liver function. Renal function showed urea 7.4 mmol/L, creatinine 112 mmol/L, eGFR 61 mL/min, which was the baseline. The C reactive protein (CRP) level was mildly raised at 19 g/L. Since the chemotherapy treatment there was a chronic normocytic normochromic anaemia, haemoglobin 82 g/L and thrombocytopenia with platelet count of 34×109/L, attributable to the cladribine chemotherapy. ANA, ANCA, complement factors 3 and 4, rheumatoid factor, immunoglobulins, cryoglobulins and a full virology screen were normal or negative. Urinalysis was negative in all parameters. Bone marrow biopsy revealed trilineage dysplastic features; no excess of blasts were seen. The myeloid lineage cells showed shift to the left and mature myeloid cells were reduced. CD34 positive cells were mildly increased in numbers.
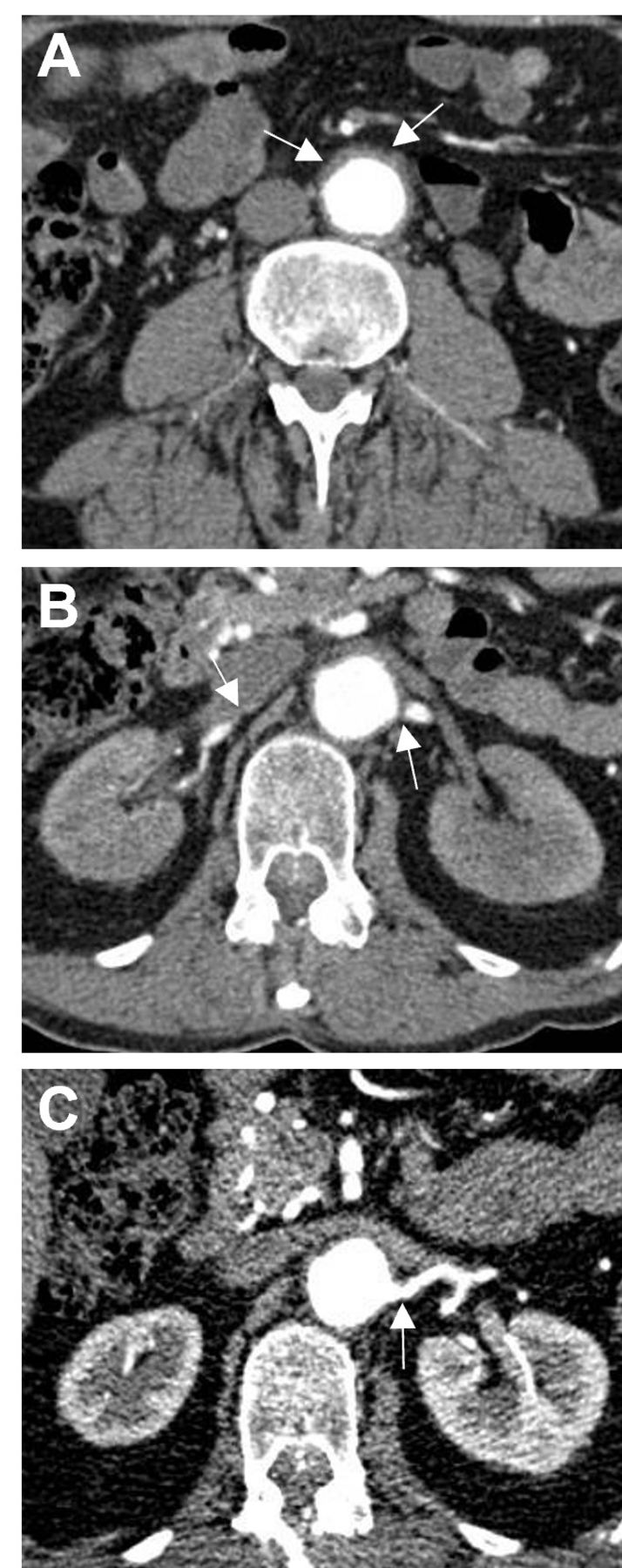
CT angiogram showing (A) diffuse wall thickening of the abdominal aorta (arrows). (B) Significant narrowing at the origin of the right and left renal arteries (arrows). (C) Marked improvement in the calibre of the left renal artery 6 months after left renal artery angioplasty.
To further investigate the aortic wall thickening, an 18F-FDG PET/CT was performed which revealed no evidence for FDG avidity in the vasculature.
Differential diagnosis
A number of factors made the diagnostic process complicated. The presence of aortic wall thickening, left subclavian and visceral artery stenoses with a raised CRP left open the diagnosis of a large vessel vasculitis. However, the absence of systemic symptoms or other signs of chronic inflammation including a normal serum albumin level made this diagnosis less likely. However, interpretation of the blood tests was complicated by the previous chemotherapy. The latter was held responsible for the normochromic normocytic anaemia and thrombocytopenia and this was confirmed following bone marrow analysis. Arterial wall thickening on a CTA scan does not necessarily equate to active inflammation and to investigate this further an 18F-FDG-PET/CT scan was performed. The lack of any evidence for FDG avidity in the arterial wall at sites of thickening or elsewhere, while not completely excluding a large vessel vasculitis, suggested there was no detectable arterial wall inflammation. As a consequence of these findings and the pre-existing diagnosis of LCH the possibility of ECD was considered. ECD is a known mimic of large vessel vasculitis. Plain radiographs of the long bones (femurs, tibia and fibula) were performed and demonstrated irregular thickening of the cortices, a marked striated appearance of the lower femora, with areas of sclerosis and patchy lucency, a classic finding in ECD (figure 2). A diagnosis of LCH/ECD overlap, with xanthogranulomatous tissue infiltration affecting multiple systems including: the long bones, central nervous system (CNS) and dura, skin, arterial tree and the retroperitoneum was subsequently made.

{kind=link}
{kind=link}
Plain radiographs showing irregular thickening of the cortices, a striated appearance of the lower femora, with areas of sclerosis and patchy lucency.
Treatment
Following a multidisciplinary team discussion involving rheumatologists, radiologists and haematologists, the treatment priority was considered to be control of the refractory hypertension. The patient underwent successful angioplasty of both the right renal artery stenosis and that involving the superior left renal artery. The arteries were balloon-dilated (to 6 mm on the left and 5 mm on the right), resulting in much improved angiographic appearances postintervention (figure 1C). Although attempts were made to cross the occluded segment involving the proximal portion of the left lower pole anomalous renal artery, these were unsuccessful. Following the procedure the blood pressure fell from 241/122 mm Hg to 130/80 mm Hg. The reduction in blood pressure was noted in the right arm, and was not measured in the left arm due to the left subclavian artery stenosis. Following the renal artery angioplasty the dose of doxazosin and bisoprolol were reduced prior to discharge. He was subsequently withdrawn from antihypertensive medication at the follow-up appointment.
Outcome and follow-up
Although blood pressure control remained satisfactory, 1 year postprocedure hypertension returned and a repeat CT angiogram scan revealed recurrent right renal artery stenosis. Angiography confirmed wide patency of the left renal artery branches and a recurrent tight stenosis of the ostium of the right renal artery. Angioplasty allowed dilatation of the artery to 6 mm with excellent antegrade flow into the right kidney. However, a small number of filling defects in the periphery of the right upper pole were consistent with thrombus. A 3-month follow-up MRA showed no restenosis and the blood pressure remains well controlled currently at 132/82.
Ongoing care is provided by haematology, dermatology and rheumatology for the ECD and LCH overlap syndrome. The BRAF V600E mutation found in 50% of ECD patients was not detected. Our patient has evidence of LCH on histology, accompanied by imaging findings pathognomonic of ECD. Coexistence of both conditions is rare but some cases have been reported.4 It has been suggested that overlap cases typically present with LCH and later progress to ECD.5 This would be in keeping with our patient’s course.
The patient remains well. A follow-up cardiac-gated 18F-FDG-PET scan excluded cardiac involvement. Ongoing issues include mild thrombocytopenia, related to the cladribine chemotherapy. In the event that the LCH relapses, interferon-α and/or other biological therapy will be considered.
Discussion
ECD is a very rare disease and considered to be a form of non-LCH, resulting from the overproduction of histiocytes.1 First identified by Jakob Erdheim, an Austrian pathologist, and William Chester, an American cardiologist, in 1930, they referred to it as ‘lipoid granulomatosis’. Dr Ronald Jaffe reported a third case in 1972, naming the condition Erdheim-Chester disease (ECD).4 The current prevalence of ECD is unknown. The disease is poorly recognised, diagnosis is complicated and hence ECD is likely under diagnosed.1 ECD has a male to female ratio of 2.4 to 1 and typically presents between 50 and 70 years of age.1 6
Although the exact aetiology of ECD is unknown, it has been proposed that proliferation of CD68+ and CD1a− histiocytes results in infiltration and accumulation in organs, resulting in tissue fibrosis.2
BRAF V600E gene deregulation has been implicated in ECD, with BRAF V600E mutations detected in 54% of those with ECD and 38% with LCH, while the mutation was absent in those with the other histiocytoses.7 The mutation results in uncontrolled generation of histiocytes via activation of the RAS/MAPK pathway.7 ECD predominantly affects the skeleton (95%), CNS (40%), skin, the retroperitoneum including kidneys (40%–50%) and the cardiorespiratory organs (40%–60%).1 3
Definitive diagnosis of ECD is through biopsy, demonstrating abnormal proliferation and accumulation of CD68+ and CD1a− histiocytes.1 In this case, biopsy was unattainable, and the diagnosis was made on circumstantial evidence. The differential diagnosis is wide and includes LCH, Paget’s disease and POEMS syndrome, haemophagocytic lymphohistiocytosis and juvenile xanthogranuloma (JXG). A bone biopsy is needed to differentiate histologically and immunophenotypically between the histiocytosis.8
Clinical manifestations
Osseous involvement is present in 95% of patients at the time of diagnosis.9 Retroperitoneal disease manifests as renal impairment, secondary to renal artery stenosis or ureteric obstruction.8 Up to 50% of patients have cardiorespiratory involvement.9 Periaortic fibrosis and arterial infiltration results in high risk of mortality, due to myocardial infarction, cardiomyopathy and valvular disease.10 CNS involvement is common, varying from periorbital changes, central diabetes insipidus, cerebellar syndromes and symptoms secondary to mass lesions.11
As in our patient, ECD may mimic vasculitis with the thoracic and abdominal aorta commonly affected (48%).12 Periarterial thickening affecting the left subclavian artery is seen in 11% of patients, as in our patient.12 Very few cases of ECD with vascular involvement have been reported and included a 70-year-old woman with ECD mimicking vasculitis.13 The patient was treated for presumed large vessel vasculitis with immunotherapy, based on imaging findings, but rapidly developed spinal column and mesenteric ischaemia and died of multiple organ failure due to septic shock.13 Histology of the large vessels and subdural area revealed ‘inflammatory changes with accumulation of foamy macrophages with CD68 +histiocytes’, specific for ECD, and the BRAF V600E mutation was present.13
The Mayo Clinic multidisciplinary Histiocytosis Working Group have developed an algorithm for initial diagnosis, including three major and four minor features, which should prompt diagnosis of histiocytic neoplasms.3 Of these features, the patient described had four of these, including DI, long bone involvement with lytic lesions, medium vessel involvement and retroperitoneal tissue involvement. For diagnosis, they recommend a full body FDG PET-CT, biopsy of FDG-avid tissue, BRAF V600 E mutation testing and staging studies.3 Ultimately, the presence of central DI with bony pain should prompt urgent consideration of ECD and LCH.3
For active disease, interferon-α is the mainstay of treatment, with 70% survival rates at 5 years.9 For those with a BRAF mutation, the BRAF inhibitor vemurafenib has been shown to be effective in the VE-Basket study, achieving 55% overall response and further research is ongoing.14 There is a role for additional biological agents.15 Anakinra has shown the best clinical response rate (50%), while tumour necrosis factor-α inhibition (infliximab or etanercept) has not proved to be effective.15
Patient’s perspective
At the beginning, symptoms started benignly and were easy to shrug off as something else. I remember early xanthelasma appearing (‘high cholesterol’) and excessive thirst first (‘dehydration’). Subsequently, my skin also started to redden in places. It very slowly worsened and holes had appeared in the skin on my head, middle back and my ear canals were weeping and gooey. This was attributed to ‘work stress’, ‘exposure to the sun’ and ‘diet’. There was a dermatological breakthrough with a diagnosis of Langerhans cell histiocytosis. ‘Mission accomplished’, my skin started clearing up with medication and my diabetes insipidus was under control. However, new adverse symptoms accumulated afterwards, causing multiple problems. These required running repairs and treatments with the help of brilliant medical support and my extraordinary wife that never gave up striving for an answer. Diagnosis, while welcome, always leaves me flat in comparison with the efforts, care and expertise of others to keep me on the road during the difficult time. I would sum the experience as: bafflement, blind alleys, uncertainty, complexity, resolve, perseverance, near misses, luck, inspiration, hope and recovery.
Learning points
Erdheim-Chester Disease (ECD) is an extremely rare condition and a form of non-Langerhans cell histiocytosis, characterised by an accumulation and infiltration of histiocytes in tissue.
ECD with vascular involvement may mimic large vessel vasculitis.
CT/MRI/PET scans, although helpful, cannot definitively differentiate ECD and large vessel vasculitis. ECD can be distinguished from large vessel vasculitis by: the presence of bony pain, classical bone radiograph findings and the presence of the BRAF V600E mutation.
Definitive diagnosis of ECD is through dedicated plain radiographs of the long bones and biopsy of affected tissue, showing abnormal proliferation and accumulation of CD68+ and CD1a− histiocytes.
The Mayo Clinic Histiocytosis Working Group algorithm provides a framework for the diagnosis of histiocytic neoplasms.
Ethics statements
Patient consent for publication
References
Footnotes
Contributors PL wrote and drafted the report. CB revised the aspects on dermatological diagnosis and management in the paper. JM oversaw the paper, created the figures and revised the final edit.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Case reports provide a valuable learning resource for the scientific community and can indicate areas of interest for future research. They should not be used in isolation to guide treatment choices or public health policy.
Competing interests None declared.
Provenance and peer review Not commissioned; externally peer reviewed.