Article Text

Statistics from Altmetric.com
Description
A 14-year-old girl presented with progressive upper and lower extremity weakness. Symptoms started 5 months prior with intermittent bilateral calf pain coincident with onset of lower extremity weakness. Initial evaluation revealed normal complete blood count, complelete metabolic panel, inflammatory markers, muscle enzymes and MRI brain and spine with/without contrast. Electromyography demonstrated an acute to subacute polyradiculitis affecting the upper and lower extremities.
She was admitted for expedited evaluation. Examination was significant for lower extremity weakness and absent reflexes. Lumbar puncture (LP) showed lymphocytic pleocytosis and markedly elevated protein. Thorough infectious, oncological and rheumatological evaluations were all unremarkable. Most notable was that CSF Neuromyelitis Optica IgG and paraneoplastic antibody panel were negative, and serum ACE was normal. She was started on intravenous immunoglobulin for empiric treatment of chronic inflammatory demyelinating polyneuropathy (CIDP).
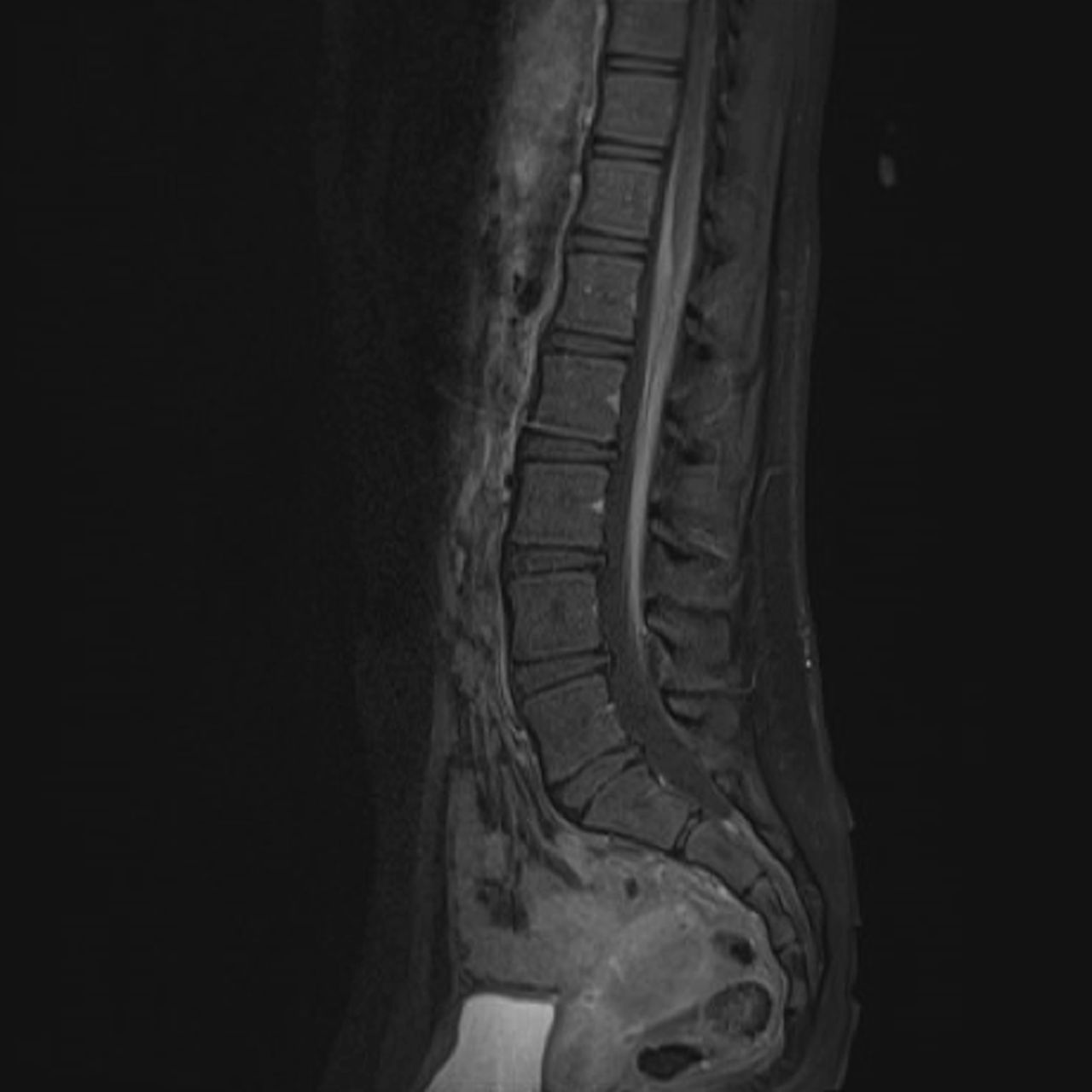
While hospitalised, she developed left eye blurred vision. Ophthalmological examination demonstrated bilateral optic nerve oedema. Repeat MRI brain showed enhancement of multiple cranial nerves (figure 1). Initial MRI spine was reinterpreted as diffuse enhancement and mild thickening of the cauda equina and cervical nerve roots (figure 2). Repeat LP had elevated opening pressure of >55 mm Hg and IgG index. She was started on intravenous methylprednisolone and acetazolamide with improvement. She was presumptively diagnosed with CIDP and discharged on a prednisone taper.
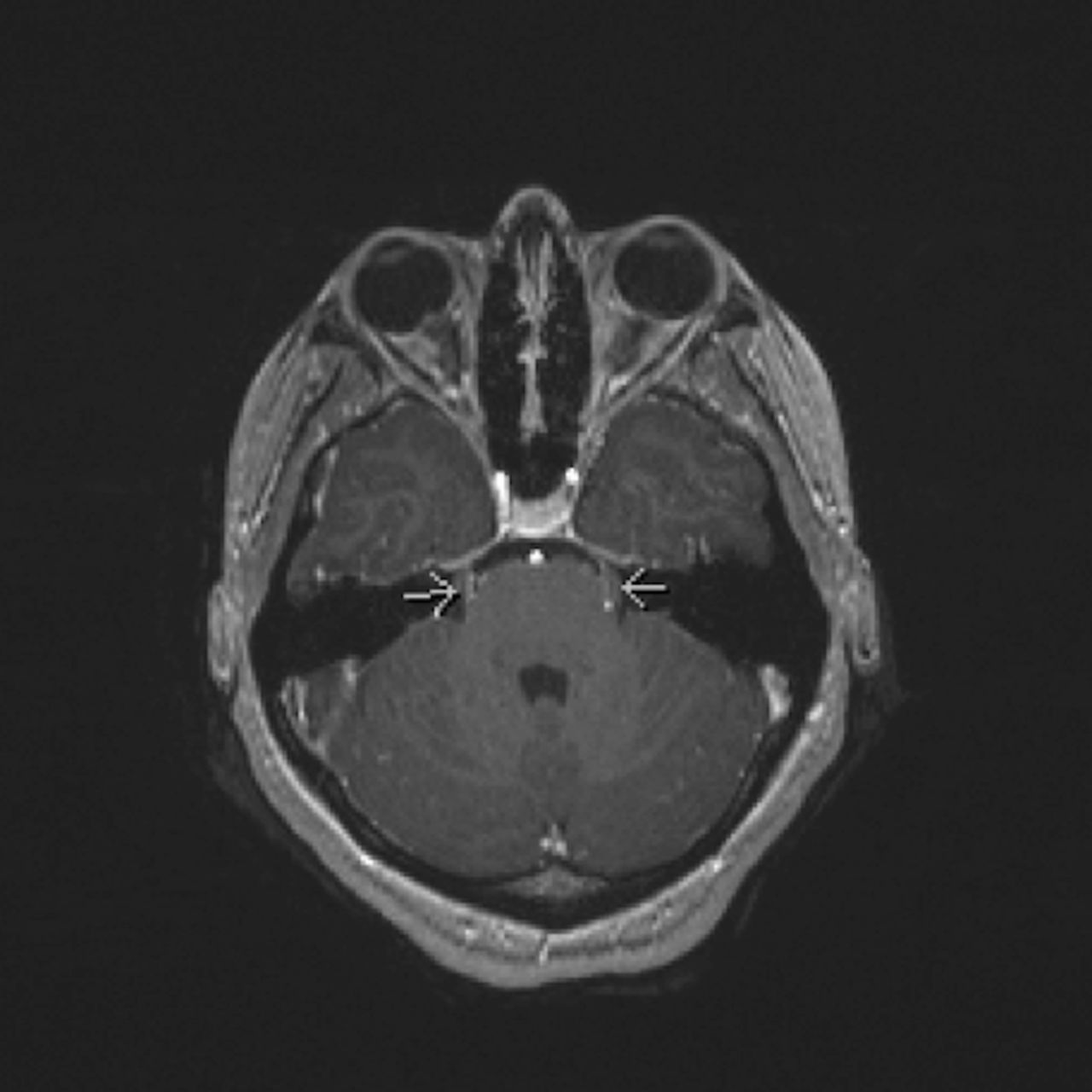
An MRI brain showing bilateral enhancement of cranial nerve V (arrows).

{kind=link}
{kind=link}
An MRI spine with contrast demonstrating diffuse enhancement and mild thickening of the cauda equina.
Within 1 week of discharge, her CSF ACE level returned elevated (3.1, reference <2.5 U/L). Given the clinical presentation, evidence of CNS inflammation by MRI and LP, and elevated CSF ACE level, she was diagnosed with possible neurosarcoidosis. She was evaluated for systemic disease which included a normal chest X-ray and negative CT of the chest/abdomen/pelvis. She was started on monthly infliximab infusions and weekly oral methotrexate with marked improvement in her symptoms, neurological examination and optic nerve oedema. In addition, she was successfully tapered off her prednisone within 6 months. A repeat MRI of her brain after 6 months of treatment showed resolution of cranial nerve enhancement and no other abnormal enhancement.
Sarcoidosis is a multisystem granulomatous inflammatory disease of unknown aetiology. Neurosarcoidosis occurs in 5%–10% of patients with systemic sarcoidosis.1 Cranial neuropathies are the most common and a classic manifestation of neurosarcoidosis. Optic nerve involvement occurs in up to 38% of patients and typically presents with visual loss and papilloedema. Elevated intracranial pressure at diagnosis is also common and has a poorer long-term prognosis.2 Isolated neurological involvement at presentation is rare and presents a diagnostic challenge. Definite diagnosis currently requires demonstration of non-caseating granuloma on tissue biopsy which is often unobtainable in patients with isolated neurosarcoidosis. This highlights the need for better, less invasive biomarkers for neurosarcoidosis. The mainstay of treatment is corticosteroids, however additional immunomodulating therapy is often required in neurosarcoidosis. Infliximab has proven to be an effective immunosuppressant for neurosarcoidosis.3
Our patient was diagnosed with possible neurosarcoidosis in the absence of being able to obtain tissue given the potential risk of a brain biopsy. Differential diagnoses considered included infection, malignancy, CIDP and other autoimmune disorders. Infection was thoroughly excluded, and she improved despite no treatment with antimicrobials. Evaluation for other autoimmune disorders was negative. While malignancy and CIDP may also respond to corticosteroids, we would not expect to see resolution with infliximab and tapering of prednisone. In addition, CIDP is a well-recognised potential side effect of infliximab treatment. Our patient’s overall clinical presentation, imaging, laboratory findings, rigorous exclusion of other causes and response to treatment are therefore supportive of possible neurosarcoidosis.
Though rare, providers need to be aware that neurosarcoidosis is a differential diagnosis for a patient presenting with cranial neuropathy, elevated ICP and/or optic neuritis. Our case highlights the high index of suspicion required for neurosarcoidosis in the absence of definitive tissue biopsy. Early and timely diagnosis is important in order to quickly initiate definitive therapy due to the high morbidity associated with the condition. More study is needed to identify better diagnostic markers and clearer treatment guidelines.
Learning points
Though rare, neurosarcoidosis should be a differential diagnosis for any patient with cranial neuropathy, elevated ICP and/or optic neuritis even if serum ACE level is normal.
When occurring in isolation, definitive diagnosis of neurosarcoidosis is challenging given biopsy may be unobtainable in these patients.
More studies are therefore needed to identify better diagnostic markers and clearer treatment guidelines for neurosarcoidosis.
Footnotes
Contributors MC and LC created the initial draft of the manuscript. LC, LKK and EYW edited the draft and subsequent versions of the manuscript. LKK and EYW cared for the patient in rheumatology clinic and provided patient updates after follow-up visits. All of the authors saw and approved the final manuscript and images prior to submission.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Patient consent Obtained.
Provenance and peer review Not commissioned; externally peer reviewed.