Article Text

Summary
We report the case of a patient with PIGT mutations who experienced recurrent aseptic meningitis 121 times over 16 years before developing paroxysmal nocturnal haemoglobinuria (PNH). Each episode was preceded by urticaria and arthralgia. After developing PNH, haemolysis occurred prior to meningitis. Flow cytometry revealed deficiency of the glycophosphatidylinositol (GPI)-anchored complement regulatory proteins, CD59 and CD55, and he was diagnosed with PNH. All the symptoms disappeared on administering eculizumab, an anti-C5 antibody. We did not detect mutation in PIGA, which is regarded as the cause of PNH. However, we detected a germ-line mutation and a somatic microdeletion in chromosome 20q including PIGT; PIGT is essential for transferring GPI anchor to the precursors of CD59 and CD55, which play important roles in complement regulation. Loss of these proteins leads to complement overactivation, causing inflammatory symptoms, including recurrent meningitis. PIGT mutations should be considered a novel pathogenesis of recurrent meningitis of unknown aetiology.
- meningitis
- haematology (incl blood transfusion)
- infection (neurology)
- immunology
- neurology
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Background
Recurrent aseptic meningitis is a rare disease which was first described in 1944 by the French neurologist Pierre Mollaret.1 Progress in biochemical and genetic analysis enabled identification of new causes of recurrent meningitis, such as herpesvirus infection, autoimmune or autoinflammatory diseases.2–5 However, the remaining aetiologies are still unknown. Here, we report the case of a patient with PIGT mutations who had experienced recurrent aseptic meningitis, paroxysmal nocturnal haemoglobinuria (PNH) and other inflammatory symptoms.
Case presentation
A 71-year-old man had experienced a total of 121 episodes of recurrent aseptic meningitis that first presented 18 years ago (figure 1). Prior to the episodes of meningitis, he had a history of recurrent urticaria and arthralgia since he was 30 years old. At the age of 53, he experienced aseptic meningitis several times a year. Low-grade fever, general fatigue, arthralgia and urticaria preceded the episodes, and high fever and headache ensued the next day. White blood cell count and C reactive protein levels were normal or mildly elevated during the meningitis period (table 1). The test results for various autoantibodies were negative. Examination of the lumbar cerebrospinal fluid demonstrated >1000 polymorphonuclear leucocytes/mm3. No bacteria, fungi or mycobacteria were cultured. PCR tests were negative for herpesviruses and enteroviruses. A biopsy from urticarial lesion showed neutrophil infiltration.
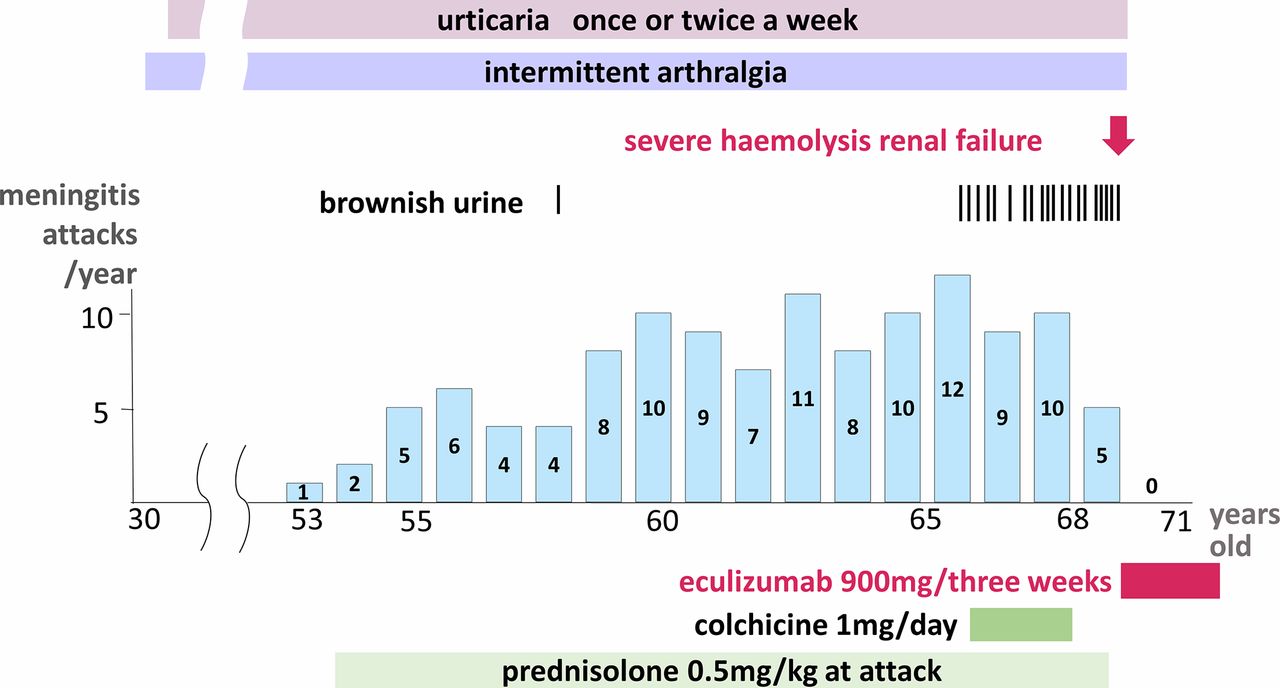
Clinical course of the patient. The number of meningitis attacks is based on the medical record calculated by calendar year. The patient had a history of urticaria and arthralgia since he was 30 years old. At the age of 53, he developed aseptic meningitis. Meningitis attacks gradually increased and sometimes accompanied by brownish urine. Finally, he developed severe haemolysis with acute renal failure. All the symptoms, including meningitis, completely disappeared soon after the administration of eculizumab.
Laboratory findings at each clinical phase
Symptoms of meningitis recovered spontaneously 7–10 days after its onset. Corticosteroid administration shortened the duration, but was not effective in preventing the attacks. We also used colchicine as a diagnostic and therapeutic agent, in case an autoinflammatory disease, such as familial Mediterranean fever (FMF), was associated with the recurrent meningitis. Genetic analysis revealed no mutations in MEFV, MVK, TNFRSF1A, NLRP3 and NLRP12.
Meningitis started occurring more frequently and was accompanied by sudden brownish urine and occasionally by altered consciousness, which lasted for several days. At the age of 68, the patient developed severe haemolysis. Haemoglobin declined to 8.2 g/dL and lactate dehydrogenase elevated up to 3004 IU/L. He also developed acute renal failure because of the deposition of haemosiderin in renal tubules. Serum creatinine increased from 2.8 to 8.65 mg/dL. He required temporary haemodialysis for a month until his condition improved.
Investigations
A bone marrow study revealed no sign of aplasia or hypoplasia. Flow cytometric analysis of both erythrocytes and granulocytes revealed deficiency of CD59 and CD55, glycophosphatidylinositol (GPI)-anchored protein, and he was diagnosed with PNH in addition to recurrent meningitis (figure 2A).

{kind=link}
{kind=link}
Flow cytometric analysis of blood cells and sequencing of genes involved in glycophosphatidylinositol (GPI) anchor synthesis. The expression of CD59 and CD55 on red blood cells and granulocytes of the patient shows a mosaic of cells with normal expression of GPI-anchored proteins and those with reduced expression of GPI-anchored proteins (A). Target exome sequence covering GPI biosynthesis genes of genomic DNA from sorted GPI+ and GPI− granulocytes reveals a nonsense mutation in a germ-line allele (B) and a somatic 18 Mbp deletion in chromosome 20q (C) including PIGT.
Genetic analysis was performed after obtaining approval from the ethical committee and the patient’s informed consent. The analysis of the genes involved in GPI anchor synthesis showed no mutations in PIGA, but revealed a nonsense germ-line mutation (c.250G>T, p.E84X), and single-nucleotide polymorphism array analysis revealed a somatic 18 Mbp deletion in chromosome 20q including PIGT (figure 2B, C).
Treatment
The patient was administered eculizumab, an anti-C5 antibody, immediately after the diagnosis.
Outcome and follow-up
All the symptoms, such as urticaria, arthralgia, headache and haemolysis, completely disappeared due to regulated administration of eculizumab. The remission has lasted for 3 years as of now.
Discussion
Recurrent aseptic meningitis is a rare disease with various aetiologies, such as viral infection commonly caused by herpes simplex virus, drug hypersensitivity, intracranial tumours or collagen disease; however, the remaining aetiologies are still unknown.1–3
Our patient had experienced recurrent urticaria and arthralgia for more than 30 years prior to the onset of recurrent aseptic meningitis, which occurred 121 times over 16 years. Repeated examination revealed no sign of infection, tumour or autoimmune diseases. Thus, we initially believed that the symptoms were caused by an autoinflammatory disease, such as FMF, which is accompanied by recurrent meningitis.4 5 However, the detailed investigations did not suggest any known autoinflammatory diseases.
The patient developed PNH at the age of 68; PNH is an acquired haemolytic anaemia caused by a somatic mutation of PIGA gene located in X chromosome of haematopoietic stem cells. The encoded protein, phosphatidylinositol glycan anchor biosynthesis class A (PIGA), is essential for the biosynthesis of GPI, and loss of PIGA function leads to defects in GPI-anchored proteins, including complement regulatory proteins.6 7 Consequently, haemolysis in PNH is caused by the uncontrolled complement activation.
In 2013, a case of PNH caused by a germ-line mutation and a somatic mutation in PIGT was reported.8 This patient exhibited severe urticaria and joint pain long before developing PNH, and also developed severe bowel inflammatory disease after the onset of PNH. This patient harboured a splice-site mutation in one germ-line allele and a somatic 8 Mbp deletion, including the entire PIGT. Our patient harboured similar mutations in PIGT and exhibited recurrent aseptic meningitis in addition to inflammatory symptoms. Both cases responded to eculizumab.9 The neutrophil predominancy in CSF and skin tissue of urticaria in our patient is notable because complement activation fragment C5a can activate neutrophils.10 It is also noteworthy that the unusual symptoms similar to those of auto-inflammatory syndromes, such as chronic urticaria, joint pain or recurrent meningitis, lasted over more than 10 years before development of PNH. These unique inflammatory symptoms and long clinical courses are common characteristics of these two cases with PIGT mutations.
Various proteins, such as enzymes, receptors, adhesion molecules and complement regulatory proteins, are anchored to the plasma membrane via GPI and play a crucial role in embryogenesis, neurodevelopmental processes and the immune system.11 12 Twenty-two PIG genes are involved in the biosynthesis and protein attachment of GPI.12 Inherited deficiency of GPI anchor leads to reduced expression or structural change of GPI-anchored proteins such as alkaline phosphatase. PIGV or PIGM mutations are known to cause epilepsy, dysmorphism and mental retardation.13 14 In addition, inherited PIGT deficiency causes skeletal anomalies but not inflammation.15
GPI-anchored protein deficiency in PIGT-PNH is caused by a germ-line mutation in PIGT gene on chromosome 20q in combination with a somatic deletion of the entire PIGT gene in haematopoietic stem cells. The GPI-anchored proteins CD59 and CD55 play essential roles in the immune system by regulating the complement pathway; PIGA catalyses the first step of the GPI-anchor synthesis, while PIGT is involved in transferring GPI to proteins.11 Both PIGA and PIGT malfunctions can induce PNH, but the mechanism of other inflammatory symptoms, including meningitis, observed in cases with PIGT, but not in PIGA mutations is unclear. Mevorach et al reported on the paediatric patients with recurrent demyelinating neuropathy with chronic haemolysis attributed to homozygote CD59 deficiency. They were all successfully treated by eculizumab.16 It is remarkable that both the deficit of GPI-anchored proteins CD59 and the lack of the mechanism transferring GPI to proteins observed in our case lead to similar recurrent inflammatory symptoms. Further studies on GPI anchor synthesis are necessary to clarify the mechanisms underlying these inflammatory symptoms.
Another distinctive feature of the present case is the long interval between the inflammatory symptoms and haemolysis. In typical PNH, haematopoietic stem cells with PIGA mutation are thought to be selected by autoimmunity, which leads to clonal expansion of PIGA mutant cells in the bone marrow.17 The mechanism of clonal expansion of PIGT mutant stem cells in the bone marrow may differ from those for PIGA mutant cells. PIGT-PNH clonal expansion in haematopoietic stem cells might be restricted in granulocytes or monocytes in early stage, which may involve in preceded inflammatory symptoms. In late stage, PIGT-PNH clone may expand in erythrocytes gradually, and finally it leads to haemolysis. This might be relevant to the atypical long clinical course. Further studies are required to elucidate the role of PIGT mutations.
In conclusion, we report the case of a patient with PIGT mutations who had experienced recurrent aseptic meningitis, PNH and other inflammatory symptoms, and was successfully treated with eculizumab. PIGT mutations should be considered a novel pathogenesis of recurrent meningitis of unknown aetiology.
Learning points
Inflammatory symptoms, such as chronic urticaria, joint pain and recurrent meningitis, and atypical long clinical course are common characteristics of paroxysmal nocturnal haemoglobinuria with PIGT mutations.
Loss of glycophosphatidylinositol-anchor function leads to complement overactivation, which may cause inflammatory symptoms, including recurrent meningitis.
The aforementioned symptoms disappear on administration of eculizumab, an anti-C5 antibody.
References
Footnotes
Contributors MK conceived the case report, collected the data, reviewed the literature, prepared the first manuscript draft and made critical revisions to the article. YM contributed to the gene analysis and interpretation and reviewed the literature and the manuscript. TK contributed to the gene analysis and interpretation and reviewed the manuscript. NK also contributed to the conception of the case report and reviewed the manuscript. All authors approved the final version of the manuscript.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Patient consent Obtained.
Provenance and peer review Not commissioned; externally peer reviewed.