Article Text

Summary
We report a case of a middle-aged woman who initially presented with a painful solitary destructive lesion at fifth lumbar vertebra. The initial diagnosis of plasma cell neoplasm was made based on limited histological information obtained from fragmented tissue sample. Clinicopathological findings were consistent with a solitary plasmacytoma, and she was treated with definitive radiotherapy. A month after completing radiotherapy, she was found to have multiple liver lesions. Subsequent liver biopsy confirmed plasmablastic lymphoma (PBL). She was treated with multiple lines of chemo/immunotherapy regimens with limited or no response. She died of progression of liver lesions causing hepatic failure 16 months post diagnosis. Because of its rarity and heterogeneous presentations, PBL could easily be overlooked clinically and pathologically in immunocompetent patients. Diagnosis of PBL should be considered when there is coexpression of myeloma and lymphoma immune markers.
- malignant and benign haematology
- radiotherapy
- chemotherapy
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Background
Plasmablastic lymphoma (PBL) is distinct type of diffuse large B cell lymphoma (DLBCL) predominantly seen in HIV-positive patients.1 The diagnosis of PBL could be a challenge due to its overlapping characterises with those of myeloma and lymphoma. Because of its rarity, no standard management strategy has been established. A review by Morscio et al showed a median overall survival (OS) of 8 months. It also revealed HIV-negative patients have slightly better OS (11 months) compared with HIV-positive patients (10 months).2
HIV-negative PBL has been shown to affect relatively higher proportion of female patients in contrast to HIV-positive patients. The median age of HIV-negative PBL patients was 55 years.3 PBL in immunocompetent patients appeared to be more heterogeneous in terms of sites of involvement.3
We describe a HIV-negative case of PBL who initially appeared to have solitary plasmacytoma. Three months later, she was found to have stage IV PBL. She was treated with different chemotherapy and immunotherapy combinations and survived for 16 months.
Case presentation
A middle-aged previously healthy woman initially presented with 2 months history of left-sided sciatica-like pain. The pain then progressed to involve lower back. She did not have any B symptoms. The initial CT of lumbar spine showed a pathological fracture in L5. Subsequent MRI of lumbosacral spine demonstrated posterior extradural mass at L5 level with compression of L5 nerve root (figure 1). She proceeded to have laminectomy and surgical decompression. Intraoperatively fibrous organising lesion intimately associated with theca at S1 level was identified. The culture from surgical material grew Staphylococcus epidermidis, probably from contamination. Nevertheless, she was treated with antibiotic therapy for extended period.

Sagittal section of T1 MRI with Gd contrast (A) reveals an enhancing extradural soft tissue mass at the level of L5. The axial view (B) revealed compression of the left L5 nerve root.
The histopathological examination of surgical specimen revealed poorly differentiated malignancy, likely haematological origin (figure 2). Immunophenotyping showed positive staining for CD45, CD138 and vimentin and negative staining for CD20, cytokeratin and thyroid transcription factor 1 (TTF-1). The ki67 index was high (70%). No fluorescence in situ hybridization (FISH) panel was performed and the specimen was negative for Epstein-Barr virus assessed by the Epstein-Barr virus encoded RNAs (EBER) in situ hybridisation. The myeloid markers were negative and a diagnosis of plasma cell neoplasm was made. Subsequent (positron emission tomography) PET did not show any other fluorodeoxyglucose (FDG) avid areas apart from L5 lesion with a maximum standardised uptake value (SUVmax) of 11.5. Bone marrow aspirate showed no increased plasma cells, no clonality and normal cytogenetics. Capillary serum electrophoresis and serum-free light chains did not demonstrate a paraprotein or light chain excess. Full blood count (FBC), liver function including lactate dehydrogenase (LDH) level were within normal range.

Histological study of the lumbar lesion. A high-power view of the sample showing a poorly differentiated tumour composed of medium to large cells.
She received 45Gy in 25 fractions of radical radiotherapy to the L5 region. Her back pain and sciatica completely resolved after the radiotherapy.
Six weeks after completing radiotherapy (4 months since initial diagnosis), she developed upper abdominal pain and non-specific flu-like symptoms. A restaging PET demonstrated complete metabolic response on the L5 region. However, multiple new liver lesions (figure 3) were identified. In addition, a new FDG avid left internal mammary node and a bony lesion at the right scapula were noted. Her LDH level was elevated to 868 U/L (normal: 120–250). Alanine transaminase (ALT), aspartate transaminase (AST) and gamma glutamyl transferase (GGT) levels were slightly elevated. FBC was normal.
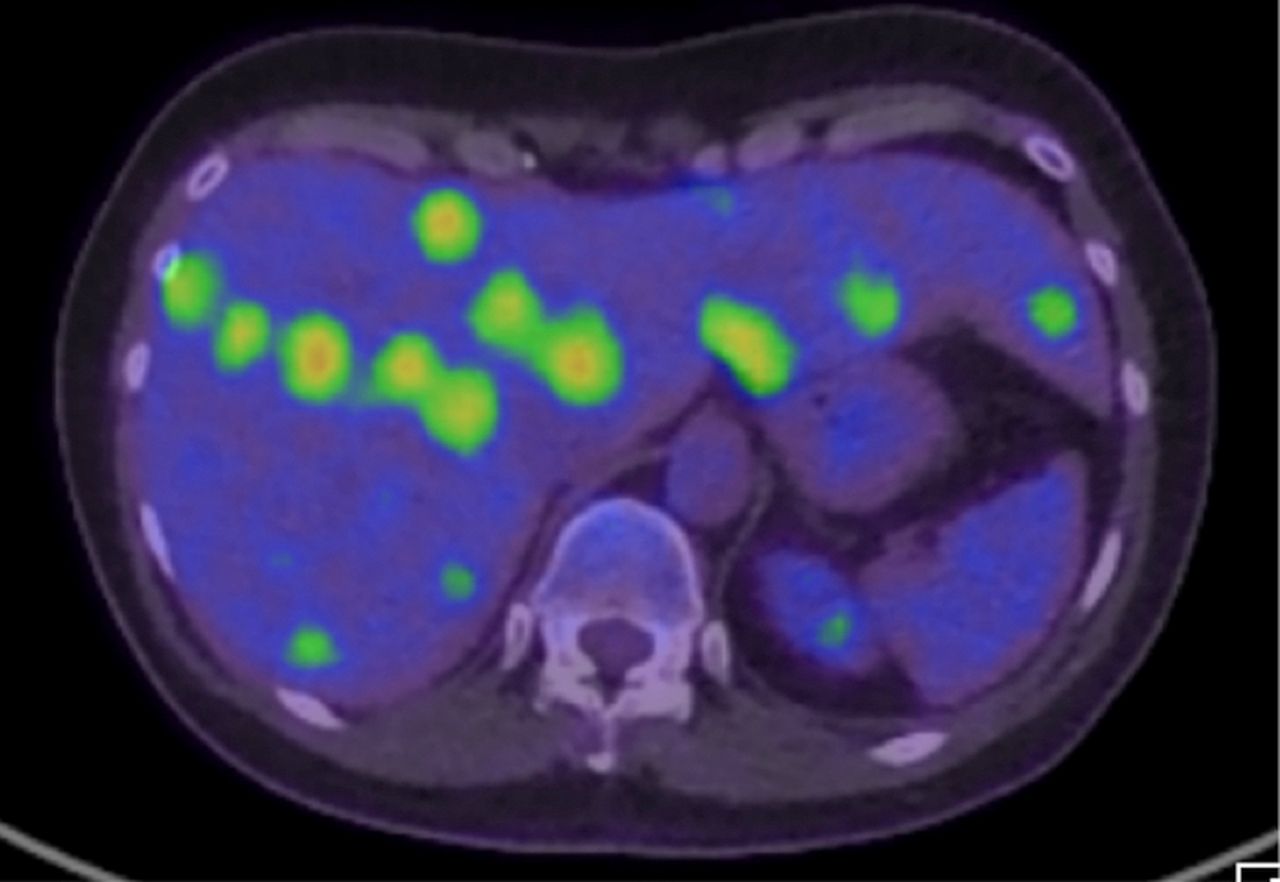
CT/positron emission tomography demonstrates multiple FDG avid liver lesions. FDG, fluorodeoxyglucose.
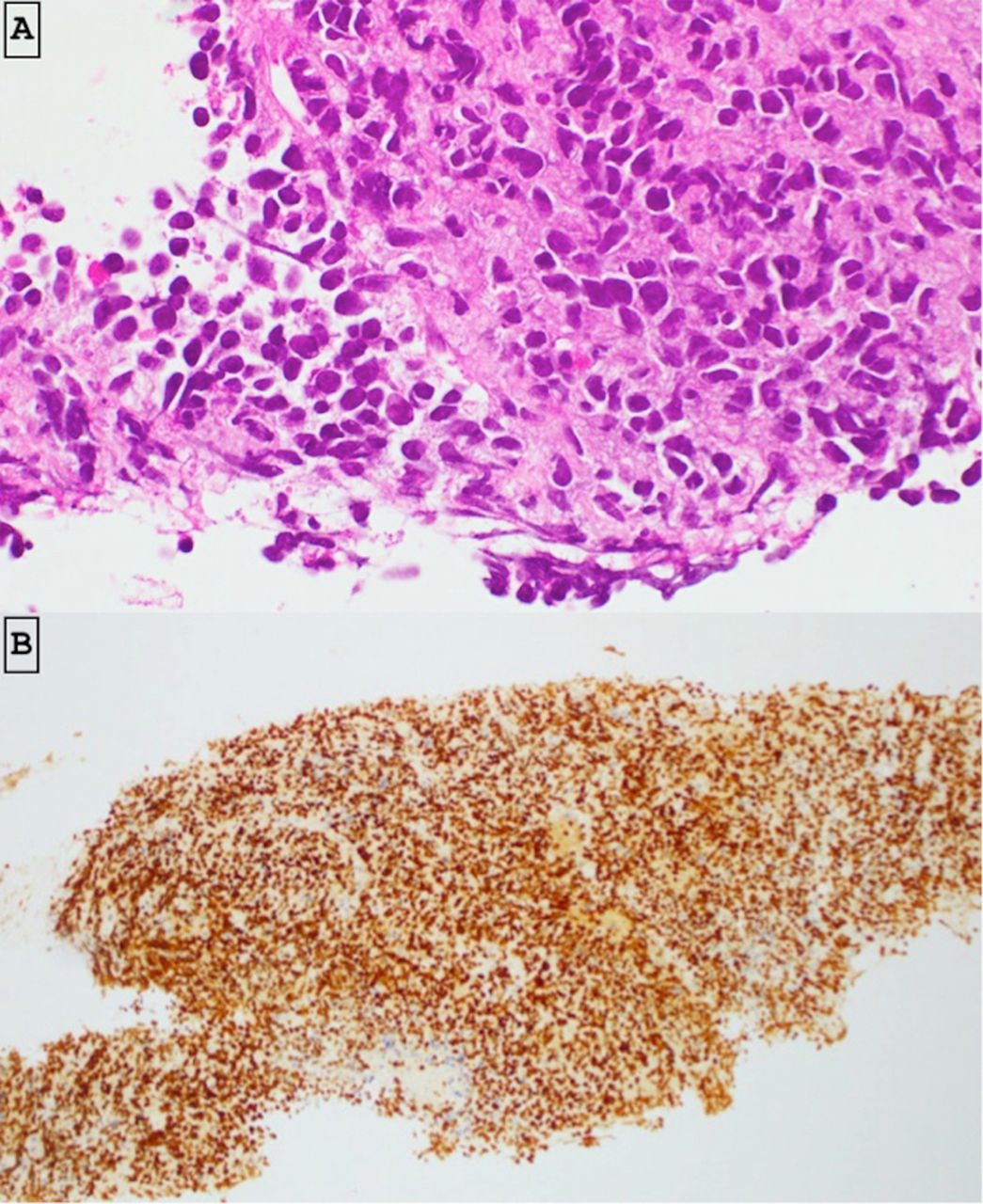
A biopsy from the liver lesion was suggestive of high-grade non-Hodgkin’s lymphoma (figure 4). The following markers showed positive immunostaining: CD45, MUM1, c-Myc, CD138, Bcl2 and Bcl6 (weak). Negative staining was noted for CD79a CD20, ALK, CD43, CD56, CD15, CD34 and chromogranin. Approximately 90% of tumour cells showed ki67 nuclear staining. Bcl2 gene rearrangement was not detected on FISH but did show loss of 3’ end of the MYC locus. This was considered as an abnormality of uncertain significance. Expert anatomical pathology opinion was that the features were consistent with a CD20 negative, aggressive B-cell lymphoma, most likely PBL. The morphological and immunological features of original biopsy taken from L5 were found to be consistent with that of the liver biopsy. The International Prognostic Index (IPI) score was 3 (increased LDH, more than one extranodal site and stage IV).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Histological and immunohistochemical study of the liver lesion. (A) Histology showed high-grade malignant tumour cells. The tumour consists of enlarged cells with hyperchromatic angulate nuclei and modest volumes of eosinophilic cytoplasm. (B) The tumour cells express positive staining for c-Myc (>80% cells).
She was started on chemotherapy regimen comprised of dose‐adjusted etoposide, prednisone, vincristine, cyclophosphamide, doxorubicin (DA-EPOCH). She also received bortezomib infusion and intrathecal cytarabine. A restaging PET performed after four cycles of chemotherapy showed progression of liver disease and SUVmax of 14.6. Subsequently, she proceeded to ifosfamide, carboplatin and etoposide (ICE) combination with bortezomib as salvage. A PET performed after two cycles of ICE showed a partial response particularly in the liver, and she proceeded to the third cycle.
After the third cycle, she presented with acute abdominal pain, fever and rapidly increasing LDH level (>5 X upper limit of normal value). She was started on steroids in view of rapid clinical progression. Subsequent PET showed some persistent disease in the liver with no definite disease progression; however, its interpretation was problematic due to 5 days of steroid therapy.
Salvage was commenced with gemcitabine and vinorelbine and a period of clinical stability was achieved. As symptoms of disease progression returned, a PET performed after five cycles confirmed progression of disease. Gemcitabine-based chemotherapy was ceased and she started on lenalidomide and dexamethasone. Unfortunately, she could not tolerate lenalidomide due to rapid deterioration of liver function associated with upper abdominal pain that likely reflected further disease progression. She received a course of palliative radiotherapy to liver to control the abdominal pain.
Investigations
See above
Differential diagnosis
Differential diagnosis consideration for PBL include a wide range of other lymphoid tumours1 4 with immunoblastic, plasmablastic or plasmacytic or immunoblastic appearances, such as DLBCL with plasmacytic differentiation, plasmablastic myeloma and solitary plasmacytoma (table 1). A comparison of clinical features, morphology and immunophenotype is listed on the following table.
Differential diagnosis of plasmablastic lymphoma (PBL)
DLBCL include a wide of clinical and morphological presentations. These tumours can be differentiated from PBL on the basis of positive immunoreactivity for B cell markers, such as CD20 and CD79a. However, both PBL and plasmablastic plasma cells neoplasms share virtually identical morphological and immunophenotype features. Hence, the distinction between these two entities must be based on clinical differences: EBV positivity is usually associated with PBL, while end-organ damage presentation (ie, renal failure, anaemia, hypercalcaemia, renal impairment) favours a diagnosis of multiple myeloma.
Treatment
See above
Outcome and follow-up
As the last resort, she was commenced on nivolumab. Following the first cycle, she rapidly progressed to develop hepatic failure due to continuing progression of her disease and passed away around 16 months after the initial diagnosis.
Discussion
PBL is a rare type of high-grade lymphoma frequently involving extranodal sites. HIV-negative PBL patients are known to present with relatively advanced clinical stage with B symptoms, and less common bone marrow involvement than in HIV- positive patients.3
Our patient initially presented like a typical solitary plasmacytoma. The fragmented histology sample taken from laminectomy procedure favoured plasma cell neoplasm. However, extensive crush artefacts and necrosis made it difficult to recognise lymphoma-related histological features. Therefore, we should highlight the importance to obtain adequate tissue samples to facilitate accurate diagnosis. After the confirmation of PBL on liver biopsy, the original sample taken from L5 lesion was compared with the liver cores. Both specimens showed identical histological and immunological features, which confirmed same pathological process.
One of the important differential diagnoses for PBL is plasmablastic myeloma. Absence of bone marrow involvement or hypercalcaemia/renal dysfunction in our patient makes plasmablastic myeloma an unlikely diagnosis.
To our knowledge, this is a first published report on HIV-negative PBL mimicking solitary plasmacytoma. In 2010, Castillo et al5 conducted a systematic review of 76 cases of HIV-negative PBL with predominately extraoral locations and reported a median survival time of 9 months and 2-year overall survival of only 10%. Liu et al6 reviewed eight cases of HIV-negative patients with PBL who underwent chemotherapy and reported complete response in seven cases.
Unfortunately, her disease rapidly progressed to involve multiple sites, predominantly liver.
There is no well-established treatment regimen for PBL due to its rarity. Cyclophosphamide, hydroxydaunorubicin, oncovin and prednisolone (CHOP) is considered as a suboptimal treatment option and the National Comprehensive Cancer Network (NCCN) guideline recommends more aggressive therapy.7 One of the treatment options for patients with HIV-associated PBL is bortezomib alone or in combination with CHOP.8 9 Another alternative is DA-EPOCH.10 Our patient was initially treated with DA-EDOCH with intrathecal cytarabine. The role of intrathecal chemotherapy as central nervous system (CNS) prophylaxis was not well studied. In view of frequent extranodal site involvement and aggressive natural history of disease, CNS prophylaxis was justified by some authors.3
Given the unfavourable outcome of PBL and expression of myeloma markers, clinicians frequently try agents that are not standard component of lymphoma management but are routinely used in the treatment of multiple myeloma. Proteasome inhibitors have been used in PBL with variable response.11–13 Unfortunately, our patient did not respond well to bortezomib/chemotherapy combination.
Our patient failed to respond with the second and third lines of treatment (ICE and gemcitabine and vinorelbine, respectively). She could not tolerate lenalidomide. Lenalidomide based-chemotherapy has been shown to achieve good response in PBL.14–16 However, lenalidomide alone was seldom used in PBL. Interestingly, there are few case reports that has described dramatic response to single-agent lenalidomide in PBL.17 18 Nivolumab is a programmed cell death protein 1 (PD-1) inhibitor that has shown anticancer activity in relapsed haematological malignancies.19 Our patient’s disease rapidly progressed after first cycle of nivolumab and she died of hepatic failure 3 weeks later.
Learning points
This is a first reported case of plasmablastic lymphoma (PBL) mimicking solitary plasmacytoma.
The case once again emphasises the need for obtaining adequate tissue sample to facilitate accurate diagnosis on the first instance.
Coexpression of myeloma and lymphoma markers should raise the suspicion of PBL in any patient.
Footnotes
Contributors RD conceived the work and wrote the article. He assisted with the literature research, image formatting for presentation in the journal, final editing and submission of the paper. LI-G was involved in the care of the patient and assisted writing the report. JA also oversaw the creation of the report and assisted in the clinical assessment of the patient. He helped in the production of the paper, literature review and final editing. JA is also responsible for the overall content as guarantor and approved the final draft.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Patient consent Not required.
Provenance and peer review Not commissioned; externally peer reviewed.