Article Text

Summary
An 85-year-old man presented with a pale appearance and generalised pruritic papules. Laboratory investigations disclosed eosinophilia, autoimmune haemolytic anaemia, mixed hyperbilirubinaemia, cholestasis and elevated serum IgG4 levels. Abdominal sonography and CT showed progressive dilatation of biliary trees, with diffuse pancreatic enlargement and a subtle capsule-like low-density rim around the pancreatic head and body. Endoscopic retrograde cholangiopancreatography found no stone-related biliary obstruction, while endoscopic transpapillary biopsy demonstrated chronic inflammation only. Nevertheless, the diagnosis of IgG4-related disease with coexisting autoimmune haemolytic anaemia was presumed. The clinical picture and laboratory abnormalities improved after administration of moderate dose of methylprednisolone.
- endoscopy
- biliary intervention
- pancreas and biliary tract
This is an open access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited, appropriate credit is given, any changes made indicated, and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/.
Statistics from Altmetric.com
Background
IgG4-related disease (IgG4-RD) is a multi-organ, fibro-inflammatory disease with various presentations. Here, we describe a case of IgG4-RD involving pancreas, biliary trees and skin, which initially came to clinical attention due to eosinophilia, elevated IgE and itchy skin rashes, mimicking an allergic reaction. Autoimmune haemolytic anaemia (AIHA) was noted during survey. To the best of our knowledge, the coexistence of IgG4-RD and AIHA has rarely been reported in the literature. This case is of great value in showing the heterogeneity of IgG4-RD.
Case presentation
An 85-year-old male veteran was referred to the Allergy, Immunology, and Rheumatology department of a tertiary referral centre for progressive, generalised pruritic reddish rashes in the neck, trunk and limbs for 6 months; there were lethargy, poor appetite and 8 kg of body weight loss. Antihistamines and topical glucocorticoids from dermatological clinics could only result in partial relief. One month before coming to our department, he was admitted in this hospital for the treatment of a community-acquired pneumonia with levofloxacin. Microbiological cultures failed to yield micro-organisms, but eosinophilia was present, which persisted despite a therapeutic trial of mebendazole; no parasites were identified. On admission, physical examination showed pale conjunctivae, mildly icteric sclera and erythematous maculopapules in the neck, limbs and trunk with excoriations; bilateral neck small lymphadenopathies were noted. Eight months earlier, he had an adenocarcinoma of the sigmoid colon, pTisN0M0, which was resected through a laparoscopic low anterior resection, and up to the present hospitalisation, no evidence of recurrence has been detected. Otherwise, he gave no pertinent travel, family, allergy or medication histories contributing to the present manifestations.
Investigations
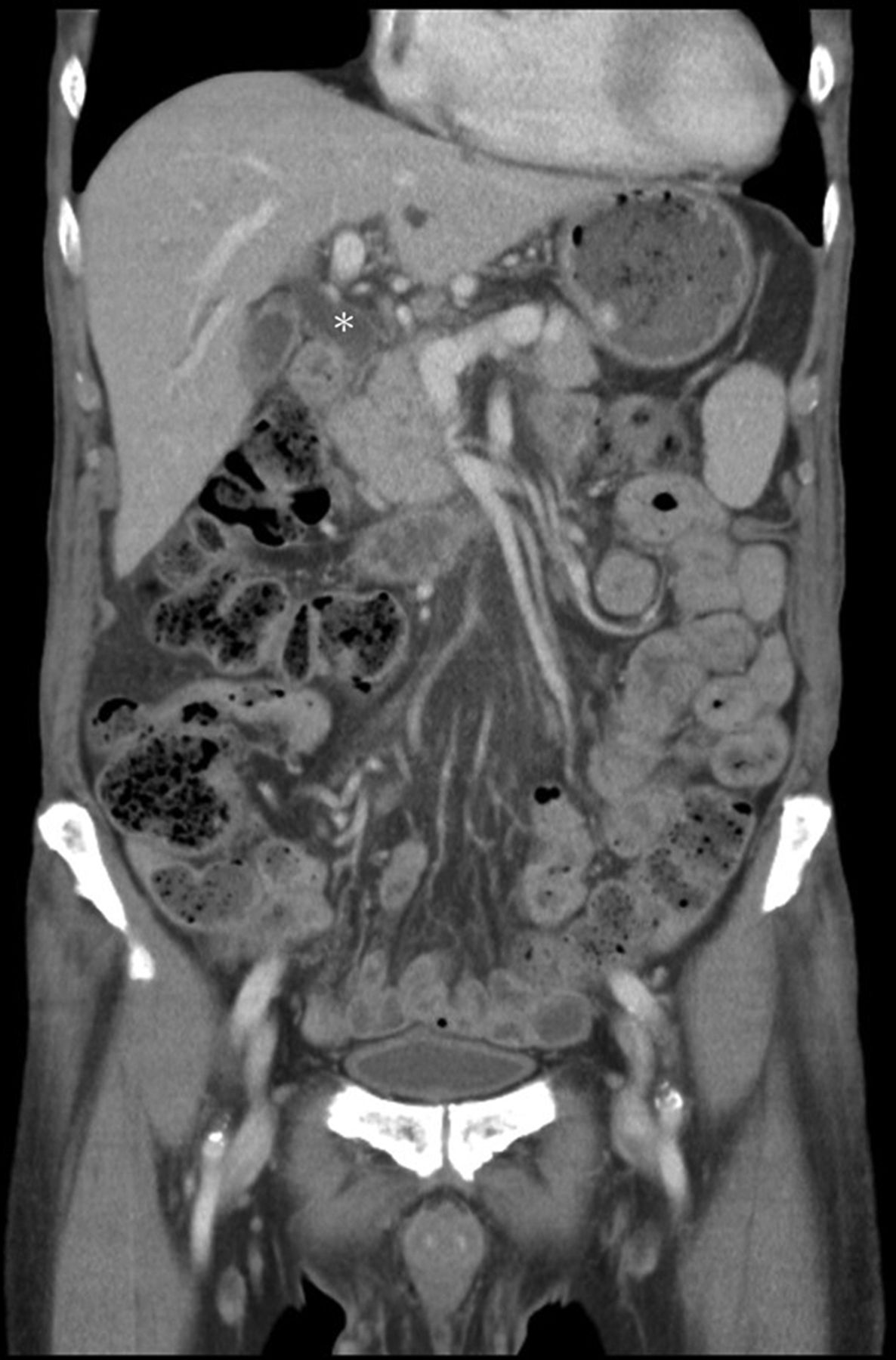
Laboratory investigations showed marked eosinophilia (3501/μL), high IgE (6095.9 IU/mL), haemoglobin of 6.4 g/dL with a mean corpuscular volume of 128.6 fL, reticulocytosis, serum aspartate transaminase of 80 U/L without simultaneous elevation of alanine transaminase, lactate dehydrogenase (LDH) of 1193 U/L, mixed hyperbilirubinaemia (total/direct bilirubin of 2.59/1.52 mg/dL), undetectable haptoglobin with a positive direct Coombs’ test and absence of parasites/ova in stool; a diagnosis of AIHA and thrombocytopenia without disseminated intravascular coagulation was made. Immunological investigations revealed low C3 and C4, but absence of antinuclear antibodies (ANAs), anti-dsDNA antibodies, antibodies against extractable nuclear antigens (ENAs) and antineutrophil cytoplasmic antibodies (ANCAs). There were high titres of lupus anticoagulant (LA), IgM anticardiolipin and anti-β2-glycoprotein-I antibodies but low titres of IgG anticardiolipin and anti-β2-glycoprotein antibodies. Strangely, there was no overt thrombosis. Bone marrow biopsy demonstrated erythroid hyperplasia. Thorough surveys failed to show evidence of infections or malignancies. Because serum alkaline phosphatase (324 U/L) and γ-glutamyl transpeptidase (147 U/L) were discrepantly high together with a mixed hyperbilirubinaemia per AIHA, abdominal sonography was carried out. Cholelithiasis with dilatation of common bile duct (CBD) as well as bilateral intrahepatic ducts was demonstrated. Contrast-enhanced CT further showed diffuse pancreatic enlargement, a subtle capsule-like low-density rim around the pancreatic head and body, and thickening of CBD walls (figures 1 and 2). Amylase (218 U/L) and lipase (127 U/L) were only mildly elevated. However, high serum IgG4 level (266 mg/dL) with increased IgG4/IgG ratio (10.9%) were noted. An endoscopic retrograde cholangiopancreatography (ERCP) showed dilatation of intrahepatic and extrahepatic bile ducts without definite intraductular filling defects (figure 3A). Because of the abnormal hepatobiliary biochemistries and cholelithiasis, an endoscopic papillotomy and balloon dilation were done to relieve the obstruction. However, there was no stone yielded after irrigations. Instead, a fibrosis of the distal CBD was demonstrated, which was presumed to be relevant to IgG4-related cholangitis (figure 3B). An endoscopic transpapillary biopsy showed duodenal tissue with chronic inflammation and scattered infiltration of IgG4-bearing plasma cells.

Axial contrast-enhanced CT showing a diffusely swollen pancreas and a surrounding low-density rim (arrowheads), compatible with IgG4-related pancreatitis. Dilatation of common bile duct (asterisk) is also present.
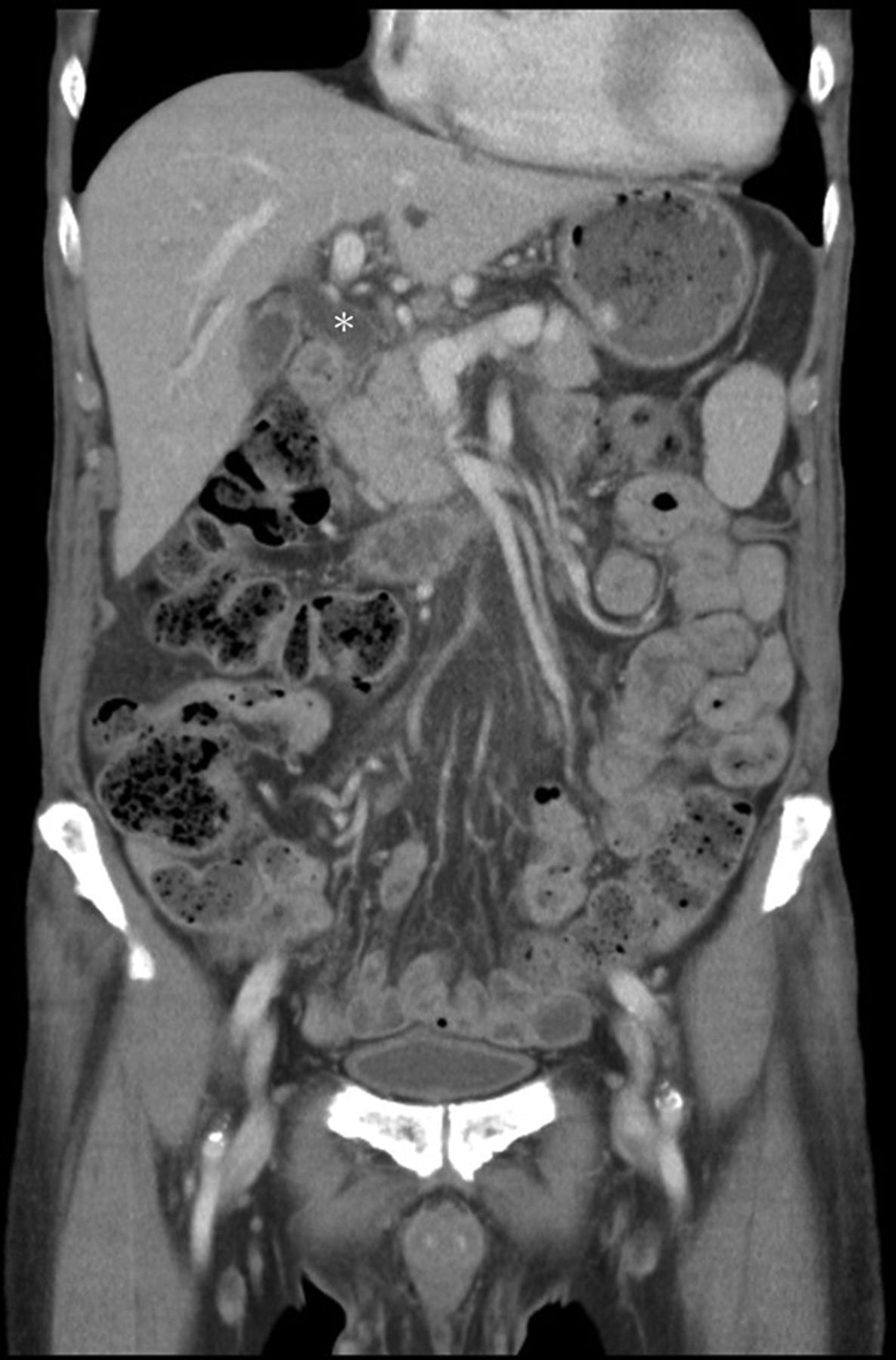
Coronal contrast-enhanced CT showing pancreatic enlargement, dilatation of common bile duct (CBD) (asterisk) and thickening of the CBD wall to 0.3 cm, a sign suggestive of inflammation and fibrosis.

{kind=link}
{kind=link}
{kind=link}
Endoscopic retrograde cholangiopancreatography showing (A) dilatation of extrahepatic and intrahepatic ducts, with stricture at distal common bile duct (CBD), and (B) fibrotic changes at the distal CBD.
Differential diagnosis
An allergic reaction first came to our mind as newly noted skin rashes accompanied by eosinophilia and elevated IgE developed. However, the patient did not have recent alternation in medications, food or housing environment; there was no parasite nor conceivable improvement of symptoms after antihelminth. Bacterial, fungal, mycobacterial and other microbial cultures failed to demonstrate any aetiology. Bone marrow biopsy to find the cause of AIHA/thrombocytopenia and high LDH did not reveal any haematological malignancies. There was also no other solid tumour. A diagnosis of systemic lupus erythematosus based on thrombocytopenia, AIHA, hypocomplementemia and antiphospholipid antibodies (APAs) was equivocal because of the senility onset and absence of ANA/anti-dsDNA antibodies. There was also no evidence to support ANCA-associated vasculitis (AAV). On the contrary, as abdominal CT showed the subtle but specific radiological signs for IgG4-related pancreatitis and related cholangitis with biliary stricture, IgG4-RD was suspected. Serum IgG4 levels and IgG4:IgG ratio were high even after 7 days of systemic glucocorticoid treatment. ERCP confirmed dilatation of biliary tracts and fibrosis of the distal CBD. Although the biopsy failed showing adequate amounts of IgG4-positive lymphoplasmacytic infiltrates, storiform fibrosis or obliterative phlebitis, IgG4-related cholangitis was still presumed because there were IgG4-related pancreatitis and fibrosis of the distal CBD without intraductular stones. Furthermore, the biopsy was done after glucocorticoid therapy, which might have inhibited the active inflammation by a large part. On the other hand, it is difficult to get deep tissues for confirming the diagnosis by an endoscopic transpapillary biopsy.1 2 Also, the eosinophilia with skin involvement is otherwise unexplainable. We did not perform skin biopsy as the rashes ameliorated after prescription of glucocorticoids and no new eruptions suitable for biopsy developed.
Treatment
Intravenous methylprednisolone 125 mg/day for 2 days and then 62.5 mg/day for 9 days were given with close monitoring of the eosinophilia and AIHA. This was then followed by a switch to prednisolone 10 mg/day after IgG4-RD had been diagnosed and AIHA been recovered. There was no blood transfusion throughout the course. The prednisolone dose was gradually reduced to 5 mg every other day 6 months thereafter.
Outcome and follow-up
The rash, eosinophilia and hyperbilirubinaemia alleviated on discharge. One month later, the haemoglobin level returned from 8.5 g/dL to 12.1 g/dL and steadily increased to 14.6 g/dL as of date. Abdominal sonography demonstrated no more dilatation of biliary tracts 3 months later. Serum IgG4 and biliary enzymes normalised 6 months later; APA and LA became negative. The patient reported improvement of appetite, a better well-being and experienced a 6 kg weight gain with a body mass index of 20.8 kg/m2.
Discussion
IgG4-RD is characterised by tumefactive and fibro-inflammatory involvement of multiple organs or tissues with lymphoplasmacytic cells, and is most frequently seen in middle-aged to elderly men. Along with dense infiltrates of IgG4-bearing plasma cells, storiform fibrosis and obliterative phlebitis are distinctive histological features in the involved tissue. However, the clinical presentations are heterogeneous, non-specific and subacute. Our patient has fatigue and weight loss with moderate eosinophilia and elevated IgE, which are all common features of IgG4-RD.3 These manifestations can also be present in a broad spectrum of diseases and hence do not suffice as clear guides. It was not until the CT demonstration of a classic autoimmune pancreatitis with probable sclerosing cholangitis that we realised IgG4-RD is the correct diagnosis. Erythematous papules, typically involving head and neck but have also been described on the trunk and limbs, are also common findings.4 Had we suspected IgG4-RD earlier, a skin biopsy might have revealed the correct diagnosis. Nevertheless, a good and rapid response to glucocorticoid treatment supports our diagnosis.
The pancreas is one of the most vulnerable organs in IgG4-RD. According to the international consensus diagnostic criteria proposed in 2011, the diagnosis can be made by a combination of parenchymal and ductal imaging, serum IgG4 levels, pancreatic histology, extrapancreatic disease and glucocorticoid responsiveness.5 The pancreatitis itself causes little abdominal pain, but may lead to secondary diabetes mellitus in half of all cases due to chronic inflammation. The most common presentation is obstructive jaundice due to concomitant IgG4-related sclerosing cholangitis, which can be classified according to cholangiographic features. Our patient presented with type 1 stricture localised to the CBD, which is most frequently associated with autoimmune pancreatitis.6
Patients with IgG4-RD are known to have autoantibodies of unknown clinical significances; ANA and rheumatoid factor present in 16%–50% and 20% of patients, respectively. Hypocomplementemia, as in this particular patient, presents in 21% of cases with IgG4-RD.7 8 The mechanisms underlying their association remain ambiguous, as IgG4 cannot bind complement effectively, and has a negligible ability to activate the classical complement pathway as compared with other IgG subclasses. Indeed, the coexistence with AIHA, which is largely dependent on antibody-complement activation, has rarely been described in the literature. AIHA has been reported associated with one case of IgG4-related pancreatitis, one case of IgG4-related sclerosing cholangitis (IgG4-SC), two cases of IgG4-related pancreatitis and IgG4-SC, one case of IgG4-related lung disease and one case of IgG4-related lymphadenopathy.9–14 All these patients share a favourable prognosis of both conditions after glucocorticoid treatment. In addition to AIHA, IgG4-RD coexists with immune-complex-mediated vasculitides including hypocomplementemic urticarial vasculitis, cutaneous leukocytoclastic vasculitis, Henoch-Schönlein purpura, cryoglobulinaemic vasculitis and AAV.15–18 These associations may be attributable to activation of IgG1 and IgG3, as high immunohistochemical reactivity of IgG4 in vessels may facilitate prominent deposition of all subclasses of IgG, or IgG4 synergy may result in other secondary autoantibodies to induce inflammation.16 19 Further researches are needed to elucidate the mechanism of antibody-complement activation in IgG4-RD.
The patient received surgery for colorectal cancer before he developed IgG4-RD. Interestingly, a recent retrospective study, showing that a history of malignancy is 2.5 times more likely in patients with IgG4-RD than in general population, has suggested that malignant neoplasm may be a predisposing factor to IgG4-RD but not vice versa.20
Thus, we emphasise the heterogeneity of IgG4-RD and wish to raise the awareness of IgG4-RD on encountering various arrays of clinical manifestations. Careful multidisciplinary correlations among clinical, pathological and often radiological findings are crucial for reaching the correct diagnosis, particularly when the initial presentations are non-specific, as seen in this particular case.
Learning points
IgG4-RD can present with skin rash, eosinophilia and high IgE, mimicking allergy.
A diagnosis of IgG4-RD depends on a high index of clinical suspicion supported by pathological and radiological evidence.
IgG4-RD is rarely associated with autoimmune haemolytic anaemia; glucocorticoid is effective for both diseases in combination.
Footnotes
Contributors K-CW took care of the patient, collected the pertinent data and wrote the manuscript. H-TL amended the manuscript, and C-YT verified all the data as well as supervised the whole study.
Funding Taipei Veterans General Hospital (V107D37-002-MY3-1).
Competing interests None declared.
Patient consent Obtained.
Provenance and peer review Not commissioned; externally peer reviewed.