Article Text

Statistics from Altmetric.com
Description
The inherited bone marrow failure syndromes (IBMFS) are a group of clinically heterogeneous disorders in which at least one hematopoietic cell lineage is significantly reduced in number.1 Dyskeratosis congenita (DC), an IBMFS caused by germline defects in telomere biology, is diagnosed by the presence of the classic clinical triad of nail dysplasia, lacy skin pigmentation and oral leukoplakia. There are clinical variants of DC, namely Hoyeraal-Hreidarsson syndrome, Revesz syndrome, and Coats plus syndrome (CpS) or cerebroretinal microangiopathy with calcification and cysts.2 CpS is a very rare disorder in which the most characteristic features are: retinal telangiectasia and exudates (Coats disease); distinctive pattern of intracranial calcification with an associated leukodystrophy and brain cysts; osteopenia, and a high risk of life-limiting gastrointestinal bleeding and portal hypertension caused by the development of vascular ectasias in the stomach, small intestine and liver.3
We report the case of a 10-year-old boy with a history of parental consanguinity, and personal history of intrauterine growth restriction since the 24th week of gestation, preterm delivery at 32 weeks, minor motor and cognitive development delay, exudative retinopathy since the age of 6 months and pathological fractures of femur and tibia. He was referred to our clinic due to thrombocytopenia and macrocytosis. Physical examination revealed decreased visual acuity, hyperpigmented reticular lesions on the neck, upper limbs and trunk, leukoplakia, and nail dystrophy (figure 1). DC was suspected. Bone marrow biopsy and aspirate revealed a hypocellular bone marrow with marked reduction of all haematopoietic series, without maturation anomalies or myelodysplasia. Liver ultrasound revealed a small liver with diffusely heterogeneous structure suggesting hepatic chronic disease, with normal Doppler study, finally FibroScan showed liver fibrosis stage F3 with no signs of portal hypertension.
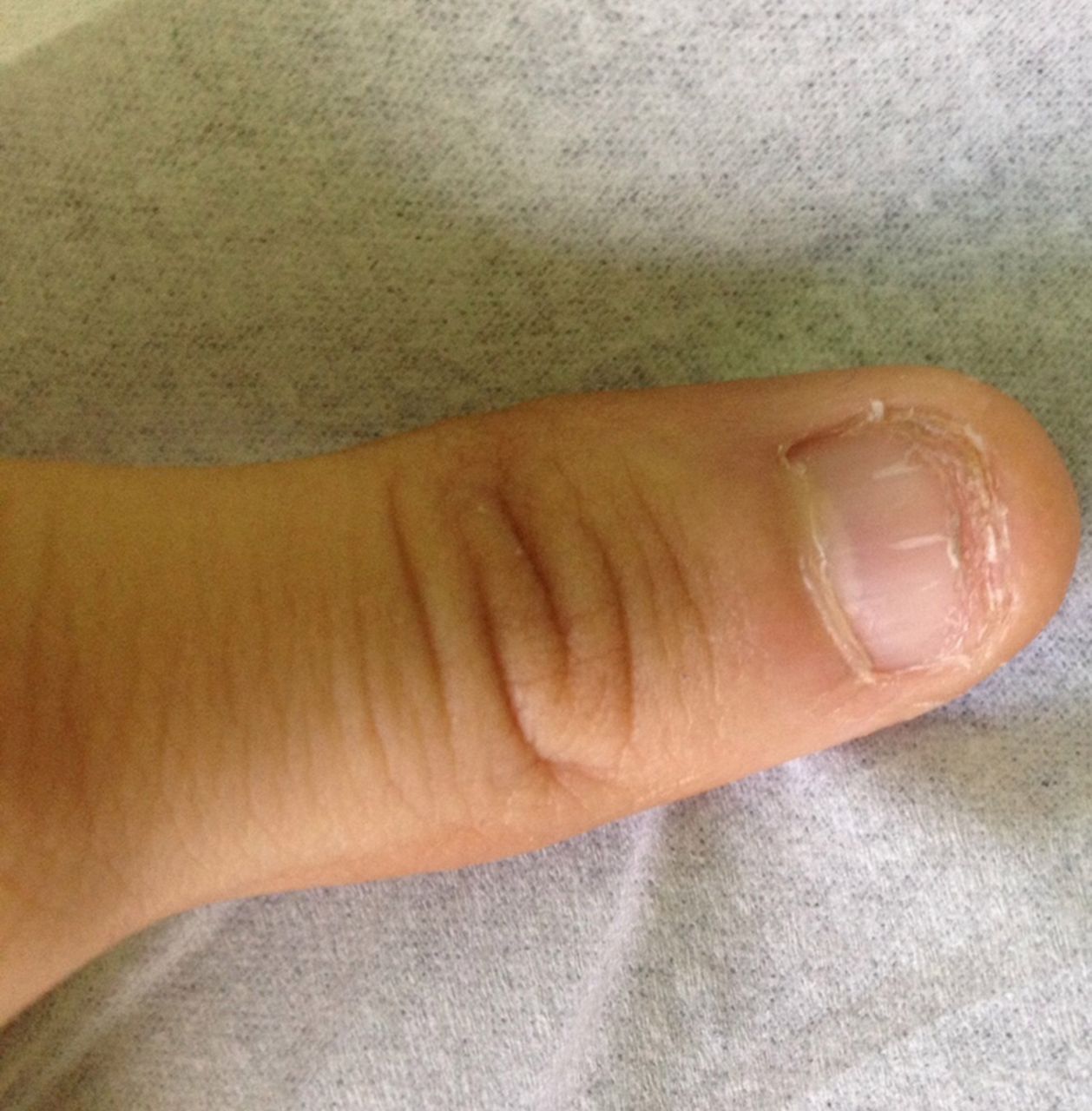
Nail dystrophy.
Due to DC suspicion, gene sequencing was performed and revealed a CTC1 mutation (sequencing of DKC1, TERC, TERT, NOP10, NHP2, TINF2, WRAP53, USB1, RTEL1, ACD and PARN was normal) suggesting CpS.
Brain MRI showed (figure 2) several asymmetric coarse calcifications in the thalami, pons, cerebellum and corticosubcortical regions of both hemispheres associated in some areas with hyperintense T2 and fluid-attenuated inversion recovery lesions of the surrounding white matter suggesting leukoencephalopathy and bilateral optic nerve atrophy. In the follow-up period, 21 months, he progressed to pancytopenia and was started on danazol, while waiting for a potential bone marrow donor.

{kind=link}
{kind=link}
Asymmetric coarse calcifications in the thalami and corticosubcortical regions of both hemispheres (SWI (susceptibility weighted imaging) sequence).
Learning points
Clinical diagnosis of dyskeratosis congenita (DC) can be challenging due to phenotypic heterogeneity and the existence of several variants of DC with overlapping symptoms, such as Coats plus syndrome (CpS).
Genetic diagnosis is the main key for the diagnosis.
In these patients with DC-like disease and overlapping symptoms, the images of brain MRI—pattern of intracranial calcification, leukodystrophy and brain cysts—may help clarify the diagnosis and identify patients with CpS.
Acknowledgments
Professor Inderjeet Dokal Centre for Genomics and Child Health Barts and The London School of Medicine London, UK
Footnotes
Contributors TP and SB were responsible for the writing and revision of the manuscript. CC and PK were responsible for revision of the manuscript.
Funding This research received no specific grant from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Patient consent Guardian consent obtained.
Provenance and peer review Not commissioned; externally peer reviewed.