Article Text

Summary
Case of cerebrofacial arteriovenous metameric syndrome (CAMS) in a 9-year-old boy is described with arteriovenous malformation simultaneously involving the brain and face, with characteristic CAMS type 1 and 2 involvement. This patient demonstrates the wide spectrum of clinical manifestations of CAMS, and in this particular case, the patient exhibits features of hypopituitarism—an association that was not previously described in the literature to our knowledge. Awareness of the underlying embryological abnormality and recognition of resultant clinical and radiological presentations are paramount for diagnosis and treatment.
- neuroimaging
- neurosurgery
- interventional radiology
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Background
This particular case of cerebrofacial arteriovenous metameric syndrome (CAMS) exhibits features of hypopituitarism, an association that was not previously described to our knowledge. Awareness of the underlying embryological abnormality and recognition of resultant clinical and radiological presentations will hopefully aid in early diagnosis and treatment in future cases.
Case presentation
A 9-year-old boy initially presented at the age of 18 months with frequent spontaneous epitaxes from the right nostril despite electrocauterisation. He was also noted to have facial asymmetry which started at infancy as a small reddish discolouration on the right cheek and gradually becoming more prominent. At the age of 5 years, he was found to have visual difficulty in the right eye during routine screening at school, which progressed to permanent monocular blindness. Pituitary dysfunction was also noted with partial growth hormone deficiency (peak growth hormone: 13.5 mu/L) and adrenocorticotropic hormone (ACTH)-cortisol deficiency (peak cortisol: 416 nmol/L), resulting in short stature and poor weight gain. He is otherwise intellectually normal and has no family history of vascular malformation or seizure.
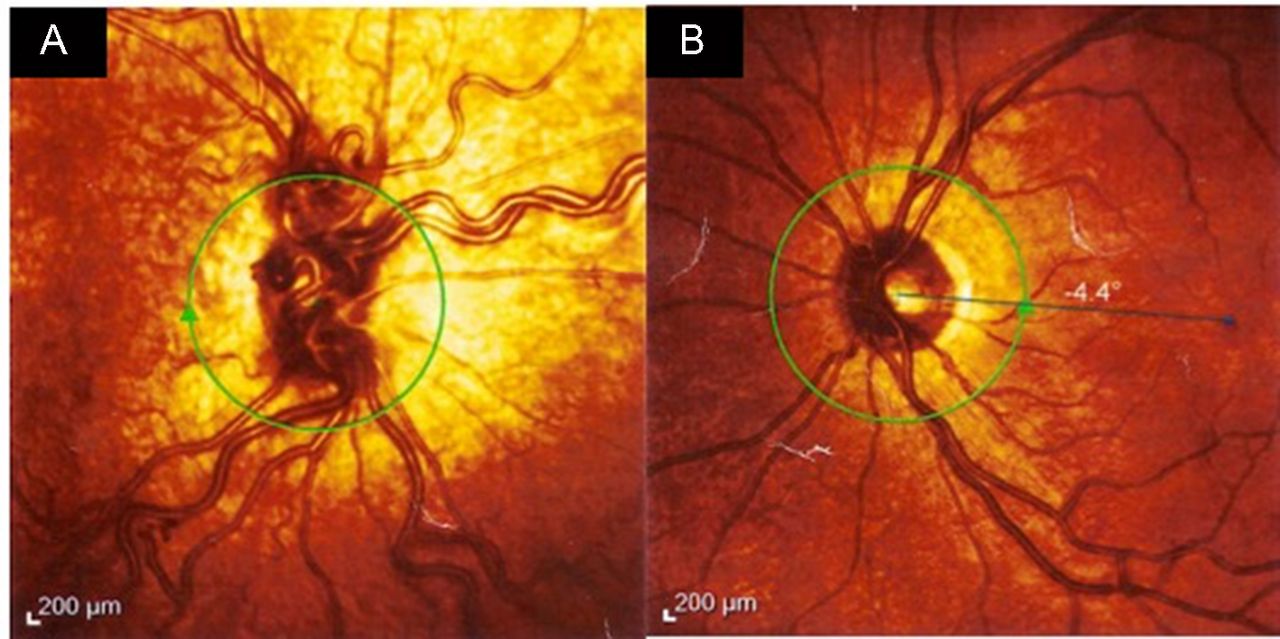
Clinical examination revealed a short but otherwise developmentally normal boy. Despite having been on steroid replacement for 4 years for hypopituitarism, he has consistently been placed at approximately the third centile for height and weight for age. There was a reddish hyperpigmentation over the V2 dermatome of the right trigeminal nerve with mild hypertrophy of the right face (figure 1). He was also completely blind in the right eye, and an afferent pupillary light defect was noted. Funduscopy of the right eye showed dilated retinal vessels as compared with the left (figure 2A,B). Anterior nasal endoscopy found prominent vessels in Little’s area and the inferior turbinate on the right side.

Our patient at age 9 years with reddish discolouration in the right V2 dermatomal distribution and mild hypertrophy of the right face.

Funduscopy of the right eye (A) at age 9 years showed dilated retinal vessels, as compared with the left eye (B).
Investigations
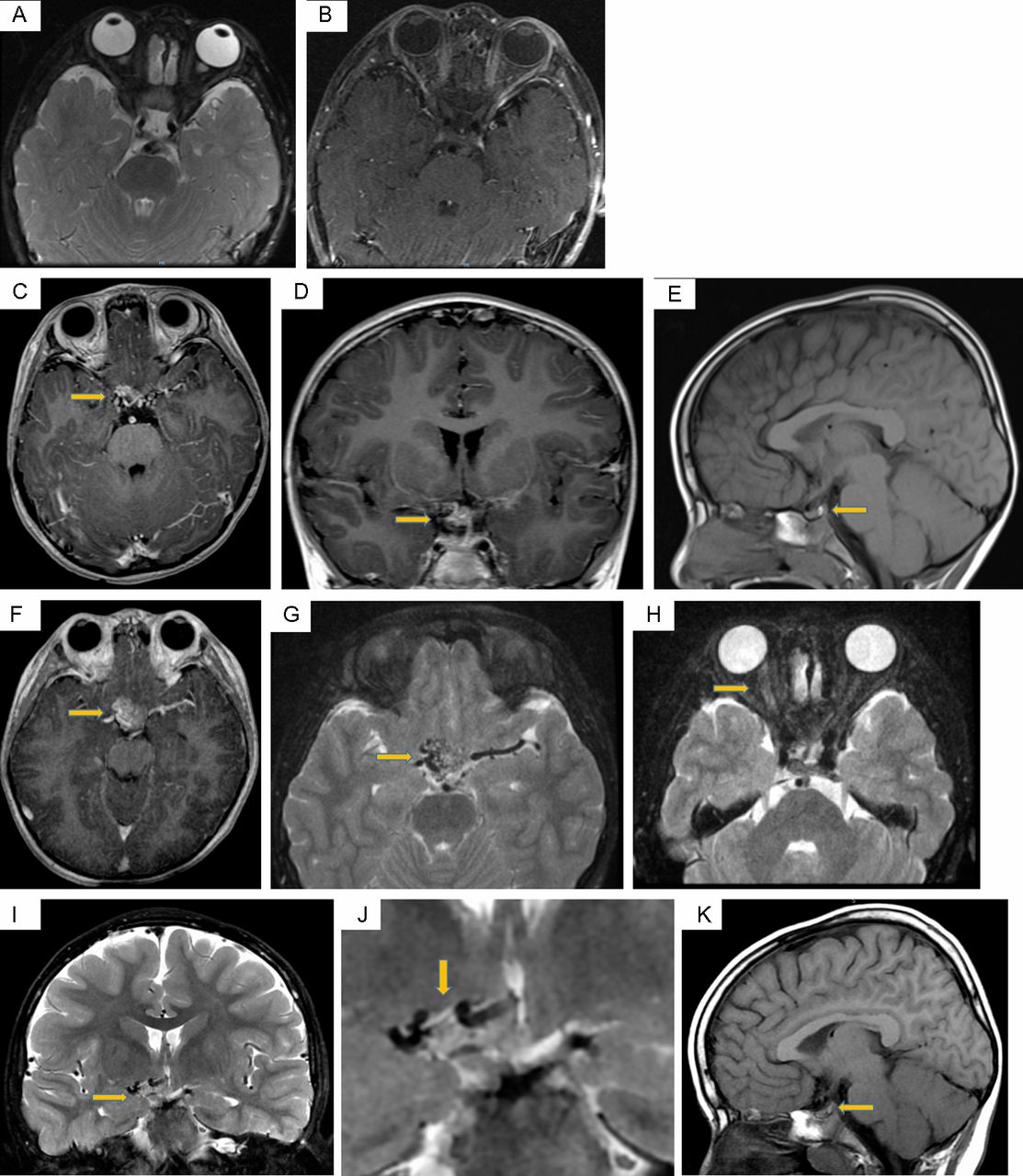
The initial MRI examination of the face at age 2 years and 3 months (figure 3A,B) was unremarkable. However, subsequent scans at ages 5 years and 3 months and 8 years and 5 months (figure 3C–K) demonstrated a gradually enlarging vascular malformation in the suprasellar region, centrally and to the right of the midline. This cluster of vessels encased the optic chiasm with involvement of the hypothalamus, and the pituitary stalk was displaced to the left. There was also extension along the right optic nerve to the level of the right orbital apex. The right optic nerve was slightly thicker than its left counterpart and was more T2W hyperintense. It also showed more avid enhancement compared with the left side on the postcontrast scan.

MRI at age 2 years and 3 months—normal MRI of the face as illustrated on axial T2W FS (A) and postcontrast T1W FS (B) images. The imaged part of the sellar/suprasellar region appears unremarkable. MRI at age 5 years and 3 months—axial (C) and coronal postcontrast T1W (D) images showed a vascular malformation in the suprasellar region, centrally and to the right of the midline. The pituitary stalk is displaced to the left but not enlarged. Sagittal T1W image (E) shows a pituitary gland of normal size, and the posterior bright spot is present. MRI at age 8 years and 5 months—axial postcontrast T1W (F), T2W FS (G,H) and coronal T2W FS (I,J) images showed interval enlargement of the vascular malformation in the suprasellar region. It encased the optic chiasm with involvement of the hypothalamus. The right optic nerve was slightly thicker than its left counterpart and was more T2W hyperintense. Sagittal T1W image (K) again demonstrates a normal-sized pituitary gland with a posterior bright spot.
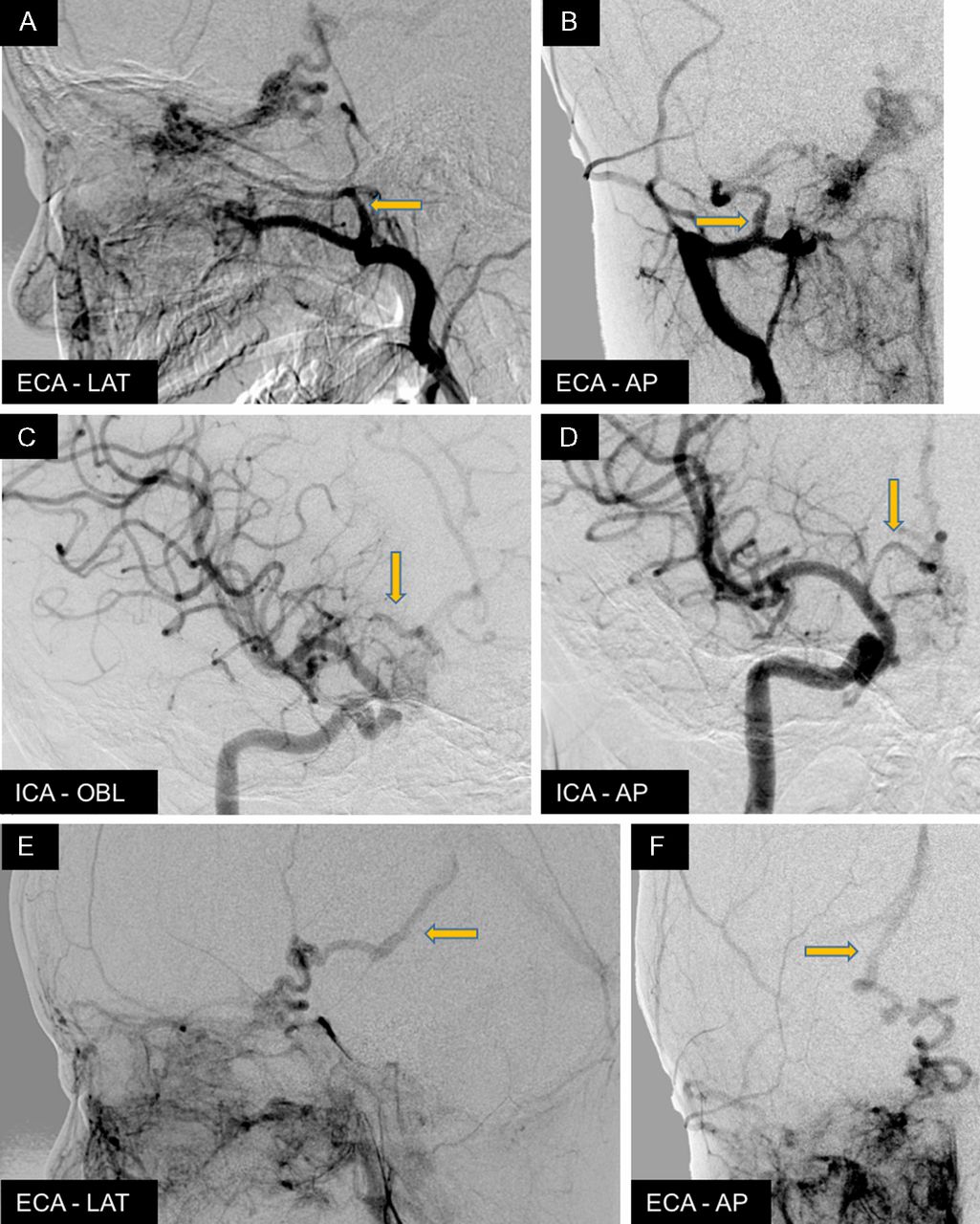
The four-vessel cerebral angiogram at age 8 years and 8 months (figure 4A–F) showed an extensive diffuse nidal-type arteriovenous malformation (AVM) centred in the region of the optic chiasm/hypothalamus, with extension of the diffuse nidal involvement along the right optic nerve to the posterior aspect of the right globe. Arterial supply pedicles appeared to come from tiny distal meningeal branches from the right middle meningeal and accessory meningeal arteries. There was also abnormal arterial supply from bilateral thalamoperforator branches from the posterior circulation and small branches from the right internal carotid artery, via the anterior superior hypophyseal and perforators from the right anterior cerebral artery/anterior communicating artery. There was also diffuse increased vascularity in the right nasomaxillary region supplied by branches of the right internal maxillary and facial arteries. This diffuse vascular blush extended into the right eye involving the preseptal and postseptal compartments, communicating with the malformation along the right optic nerve. The dominant venous drainage of the malformation was through a dilated and tortuous venous channel draining into the posterior segment of the right basal vein and into the vein of Galen and straight sinus.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Cerebral angiogram at age 8 years and 8 months. Extensive diffuse nidal-type AVM centred in the region of the optic chiasm/hypothalamus, with extension of the diffuse nidal involvement along the right optic nerve to the right globe. Arterial supply appears to come from the right middle meningeal and accessory meningeal arteries of the right external carotid artery (ECA) (A,B) and the small branches of the right anterior cerebral artery/anterior communicating artery (ACA/ACOM) of the right internal carotid artery (ICA) (C,D). Dominant venous drainage of the malformation was through a dilated and tortuous venous channel draining into the posterior segment of the right basal vein and into the vein of Galen and straight sinus (E,F). AVM, arteriovenous malformation.
Outcome and follow-up
Endovascular treatment of the intracranial AVM was considered too hazardous, and conservative management was decided unless the patient develops neurological symptoms in the future. Regular endocrine reviews are scheduled to monitor growth velocity.
Discussion
Association of facial, retinal and cerebral AVMs was first recognised by Bonnet, Dechaume and Blanc in Lyon, France in 1937 and Wyburn-Mason in London in 1943. In 2001, Bhattacharya et al suggested that the wide spectrum of phenotypic expression brought about by combination of individual vascular malformations is merely a reflection of the transverse (metameric) pattern of the underlying cerebrofacial disorder, as a result of abnormal development of neural crest or adjacent cephalic mesoderm.1 They suggested the name CAMS with a view of encompassing conditions with arterial or venous malformations that simultaneously involves the face and the brain. A previous comprehensive review by Schmidt et al found fewer than 100 cases reported in the literature.2
A new classification and diagnostic criteria were introduced, based on the metameric organisation of the central nervous system as well as the associated facial skeleton involvement derived from the same portion of the neural crest.1 Three subgroups were described: CAMS-1 represents the ‘median prosencephalic group’, involving the hypothalamus and hypophysis as the intracranial component and the nose as the facial component. CAMS-2 covers the ‘lateral prosencephalic group’, with involvement of the retina, optic tract, thalamus and occipital lobe as the intracranial component and the maxilla as the facial component. They also proposed a third group, CAMS-3 the ‘lateral rhombencephalic group’, involving the pons, cerebellum and the mandible.
Under this classification, our case cannot be strictly classified as either isolated type 1 or type 2 as this particular AVM involves overlapping territories. This, however, may be explained by Couly’s avian experiments and by extension likely same for primates, which demonstrated that regionalisation of crest and cephalic mesoderm migration are by no means absolute. Extensive insult to the prosencephalon at an early stage of development may compromise overlapped territories culminating in a mixed phenotypic expression.3
Nevertheless, the AVMs in CAMS have predilections for structures along the visual pathway: retina, optic nerve, chiasm/hypothalamus and thalamus. This is exemplified by Bhattacharya’s series of 15 cases which showed that the most common presenting symptoms is visual deterioration (in 9 of 15 cases)—as either decreased visual field or acuity.1 Other presenting complaints reported in other cases were epistaxis, headache, intracranial haemorrhage and hemiparesis.1 4 Rarer yet, there were reports of CAMS involving the thyroid, palate, pharynx and gastrointestinal tract.5 6 To our knowledge, this is the first reported case of CAMS with hypopituitarism.
A reasonable explanation for hypopituitarism in CAMS is that the presence of an AVM in the region of the optic chiasm and hypothalamus bypasses the portal and arterial circulations normally supplying the respective adenohypophysis and neurohypophysis, thereby causing ischaemia/necrosis over time. This is somewhat analogous to Sturge-Weber syndrome, where hemiatrophy of the cerebral hemispheres is probably due to altered cortical and white matter perfusion as a result of capillary venous vascular malformations.7 Another potential mechanism causing a more acute onset of pituitary dysfunction is a thromboembolic event with subsequent infarction secondary to AVM-related angiopathy. Hypopituitarism in this case is unlikely to be directly due to abnormal development of the neural crest causing defects ‘downstream’ —since only the neurohypophysis is derived from the diencephalon, and our patient suffered from partial growth hormone and ACTH-cortisol deficiencies (ie, disorder of the adenohypophysis).
With regard to disease progression, we believe that further deterioration of pituitary function is likely. Many cases of CAMS in the literature reported progressive enlargement of cerebral AVMs with neurological decline and increasing risk of intracranial haemorrhage.8 Jiarakongmun et al and Lasjaunias and Brugge previously found that the angioarchitecture of cerebral AVMs in CAMS is different to those seen in sporadic AVMs,4 8 as demonstrated by our case. Rather than the usual finding of high-flow arteriovenous fistulas or shunts, dysplastic flow-related aneurysms and dilated draining veins commonly described in sporadic AVMs, those found in CAMS, present as clusters of extensive diffuse nidal-type AVM with normal intervening brain tissues. Jiarakongmun et al and Lasjaunias and Brugge therefore postulated that cerebral AVM in CAMS follows a different natural history compared with sporadic AVMs and some other neurocutaneous disorders.
Currently, most of the intracranial AVMs in CAMS are considered incurable, although spontaneous resolution has been reported.9 Expectant management remains the prevailing standard, and treatment options such as endovascular embolisation, stereotactic radiosurgery and microsurgical resection are often reserved for those severely symptomatic patients.10 Nevertheless, awareness of the underlying embryological abnormality and recognition of resultant clinical and radiological presentations–including hypopituitarism–are paramount for diagnosis, early symptomatic treatment and possibly future consideration for endovascular intervention.
Learning points
Patients with cerebrofacial arteriovenous metameric syndrome (CAMS) may present in a wide variety of manner, including hypopituitarism.
Awareness of the underlying embryological abnormality of CAMS and its pathological involvement of the pituitary gland will aid in diagnosis.
A multidisciplinary approach is paramount for recognition and subsequent clinical management of CAMS.
Footnotes
Contributors JCN did the background research, discussed the patient’s clinical history and progress with the patient’s family, as well as wrote the bulk of the case report. CA and WL performed the four-vessel cerebral angiogram for the patient and followed up the patient in clinic.
Competing interests None declared.
Patient consent Guardian consent obtained.
Provenance and peer review Not commissioned; externally peer reviewed.