Article Text

Summary
Homozygous familial hypercholesterolaemia (HoFH) is a rare, genetic disorder of abnormally high levels of low-density lipoprotein cholesterol (LDL-C) requiring aggressive interventions to retard the evolution of atherosclerotic cardiovascular disease. We treated two brothers (ages 46 years and 47 years) with HoFH with statins, lipoproteinapheresis (LA) and the microsomal triglyceride transfer protein inhibitor lomitapide. Both brothers carried the p.Thr434Arg homozygous LDLR mutation and had childhood total cholesterol levels >700 mg/dL. Inter-LA LDL-C levels remained high; therefore, they were given escalating doses of oral lomitapide (5–10 mg/day). One brother was able to maintain LDL-C levels <70 mg/dL and stop LA. Lomitapide was well tolerated, with only an episode of headache requiring dose reduction from 40 mg/day to 20 mg/day in one patient. In two HoFH cases, lomitapide was an effective and well-tolerated adjunct therapy. Lomitapide doses required to maintain LDL-C goal levels appear to be lower in clinical practice than in clinical trials.
- lipid disorders
- endocrine system
- congenital disorders
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Background
Homozygous familial hypercholesterolaemia (HoFH) is a rare, genetic disorder of lipoprotein metabolism that is characterised by abnormally high levels of circulating low-density lipoprotein cholesterol (LDL-C).1 A recent study in Spain has revealed a prevalence of one case in 450 000 individuals, which is higher than previously expected.2 Lifelong exposure to elevated LDL-C values greatly increases the risk of atherosclerotic cardiovascular disease (ASCVD) and premature death; untreated HoFH patients often do not survive past the age of 30 years.3 In order to reduce the risk of ASCVD in patients with HoFH, it is important to correctly diagnose patients as early as possible and provide aggressive interventions to reduce the cumulative LDL-C burden.1 3
HoFH results from two mutated genes coding within one or more of the following known genes: LDLR, APOB, PCSK9,4 LDLRAP1,5 APOE6 and LIPA,7 which in turn compromises clearance of LDL-C through the low-density lipoprotein receptor (LDL-R).1 3 The most commonly mutated gene in HoFH is LDLR.8 9 Mutations on APOE can modulate lipoprotein phenotype expression and cardiovascular (CV) outcomes in patients with HoFH and are responsible for familial dysbetalipoproteinaemia.6 10 A genetic screening study of patients with HoFH in Spain showed that more than 85% of patients were carriers of LDLR mutations.2 As shown in this study, the heterogeneity of the causative mutations is thought to be largely responsible for the broad range of clinical presentations and phenotypes observed among patients with HoFH, as well as the wide range in LDL-C levels.1 The Spanish study found that more than 50% of HoFH cases in Spain do not meet classical diagnosis criteria.2 For the patients, this is important, because it can delay the diagnosis and can have important implications for their offspring.
European Atherosclerosis Society (EAS) suggests an LDL goal of <70 mg/dL in the highest risk patients.1 Importantly, these guidelines recognise that despite the large elevations in LDL-C that characterise HoFH, patients ought to be treated to the same target LDL-C levels as those with polygenic, high-risk hypercholesterolaemia. Treatment guidelines suggest that first-line therapy includes lifestyle modifications and conventional lipid-lowering therapies (LLTs) such as statins with or without ezetimibe, which may be accompanied by drugs such as bile acid sequestrants or niacin to further lower LDL-C levels.1 Because such LLTs are dependent on the LDL-R for efficacy, patients with HoFH typically experience blunted responses to these conventional LLTs. Consequently, LDL-C levels are rarely reduced to the recommended target goals.1 Therefore, lipoprotein apheresis (LA) is recommended as a treatment option for most patients with HoFH.1 A single LA treatment can reduce serum LDL-C levels by between 50% and 70%1; however, LDL-C levels rebound rapidly after treatment, returning to 50%–90% of preapheresis levels after 4 days and 14 days, respectively.11 In the experience of our clinic in Spain, LDL-C apheresis resulted in 69% decreases in LDL-C levels in patients with HoFH and also resulted in resolution of CV symptoms.12 LA is typically repeated every 1–2 weeks,1 but even with this frequency of treatment, LDL-C levels are not generally maintained within the required target range, and time-averaged LDL-C levels are usually not at goal. In fact, for patients with HoFH, recommendations suggest weekly LA regimens for CV outcomes prevention. However, for some patients with familial hypercholesterolaemia (FH), LA causes unmanageable loss of time and working days, problems with attending treatment centres and difficulties in achieving vein access for less experienced staffs. Patients consider LA sessions to be uncomfortable and incompatible with employment.10
The limitations of conventional LLTs and LA in patients with HoFH have led researchers to seek further alternatives. Lomitapide, an oral inhibitor of the microsomal triglyceride transfer protein (MTP), has been approved as an adjunct to conventional LLTs with or without LA for HoFH (with or without genetic confirmations) in patients aged ≥18 years.13 Lomitapide is not currently indicated for paediatric patients.
MTP is an enzyme that both stabilises apolipoprotein B (apoB) during its translation in the rough endoplasmic reticulum and transfers triglycerides (TGs) and phospholipids onto apoB, thereby playing a critical role in the assembly and secretion of apoB-containing lipoproteins in the liver and intestines.14 By inhibiting MTP, lomitapide induces post-translational degradation of apoB, thereby reducing the secretion of the very low-density lipoprotein and chylomicrons from the liver and intestines, respectively, resulting in lower serum cholesterol and TG levels.14
In an open-label, dose-escalation trial of 29 patients with HoFH, lomitapide (5–60 mg/day) was added to standard of care including LA.15 Of the 23 patients who completed treatment (median lomitapide dose, 40 mg/day), mean LDL-C levels were reduced by 50% at 26 weeks. The most common adverse events (AEs) reported were gastrointestinal (mitigated by adherence to a low-fat diet (ie, supplying <20% of total energy from fat)), accumulation of liver fat and elevations in alanine aminotransferase levels. These AEs can be managed with dose adjustments.
For healthcare practitioners to adopt updated treatment guidelines and novel treatment options, it is helpful to observe real-world patient experience. In this paper, we examine two ‘real-world’ cases of lomitapide use in brothers with HoFH and consider the factors that led to initiation and adaptation of therapy.
Case presentation
Patient 1
Patient 1 is a 47-year-old non-smoking male diagnosed with HoFH at the age of 8 years. At diagnosis, the patient had a total cholesterol (TC) level of 800 mg/dL, TGs at 90 mg/dL, high-density lipoprotein cholesterol levels at 34 mg/dL and LDL-C of 747 mg/dL. Xanthomas were evident on the elbows, knees and ankles. He had been treated with statins for more than 25 years and with adjunctive ezetimibe for the last 10 years.
His father had TC values >400 mg/dL and died at the age of 37 years from acute myocardial infarction. His mother has ongoing hypercholesterolaemia, which is managed with statins with TC 220 mg/dL. She has experienced no CV events to date. The patient’s brother also has HoFH and is documented as patient 2 below.
A genetic analysis confirmed the presence of the p.Thr434Arg homozygous LDLR mutation.
At the age of 21 years, the patient developed mesangial IgA glomerulonephritis that resulted in renal insufficiency requiring a kidney transplant. This graft was rejected 3 years later, and a second transplant was conducted. Glomerular filtration rate was 47 mL/min/1.73 m2, and the patient was treated with mycophenolate mofetil to suppress graft rejection.
At the age of 39 years, the patient developed coronary heart disease and underwent three-vessel coronary artery bypass graft. In the same year, an aortic abdominal aneurism occurred with plaques in the carotid artery in the absence of stenosis.
At this point, the patient had a TC level of 220 mg/dL, TG of 97 mg/dL, HDL-C levels of 32 mg/dL and LDL-C of 168 mg/dL. ApoB levels were 140 mg/dL, and Lp(a) was 60 mg/dL. The patient was being treated with atorvastatin 80 mg/day plus ezetimibe 10 mg/day. Due to high CV risk, a weekly LA was offered but was declined by the patient, who preferred to adopt a biweekly regimen. The patient was commenced on biweekly apheresis in 2014. Since the application of apheresis, there have been no additional coronary events. At this point, the patient’s weight was 80 kg, his height was 184 cm and had an abdominal circumference of 93 cm and blood pressure at 138/76 mm Hg.
Patient 2
Patient 2 is a 46-year-old male, diagnosed with HoFH at the age of 6 years (LDL-C at diagnosis was 829 mg/dL). The patient is the brother of patient 1. He carries the homozygous p.Thr434Arg mutations. He has xanthomas on the knees and Achilles’ tendon. The patient’s untreated baseline lipid levels were: TC 867 mg/dL, LDL-C 829 mg/dL, HDL-C 26 mg/dL, TG 58 mg/dL and apoB 300 mg/dL. The patient was initially treated with atorvastatin 80 mg/day plus ezetimibe 10 mg/day.
The patient underwent an episode of tuberculosis pericarditis with complete resolution at the age of 30 years. At the age of 34 years, he underwent triple coronary bypass due to coronary atherosclerosis. At the age of 40 years, the patient underwent endarterectomy of right carotid artery due to complete stenosis. In 2012, he commenced on biweekly lipoprotein apheresis. During this time, mean interval LDL-C levels were 152 mg/dL (figure 3, table 2). The medical team offered to decrease the LA interval to weekly for better control of LDL-C, but the patient declined this regimen.
Treatment
Patient 1
Despite successful application of apheresis therapy with complete regression of xanthomas, mean interval LDL-C levels remained at 128 mg/dL (figure 1, table 1). Interapheresis LDL-C levels were higher than the desired <70 mg/dL, and angiographic studies revealed an atherosclerotic plaque in the renal artery that could affect viability of a renal transplant. These factors led the medical team to add lomitapide to the patient’s treatment regimen. Importantly, the patient was taking three drugs with possible contraindications for lomitapide. The patient was receiving atorvastatin and amlodipine, both of which share the hepatic cytochrome CYP3A4 elimination route with lomitapide. The patient was also receiving tacrolimus, which may have required a dose reduction as it is a P-glycoprotein substrate and can increase exposure in the presence of lomitapide. Multimodal therapy was important for this patient because lomitapide alone cannot adequately control lipoprotein(a) (Lp(a)) levels.
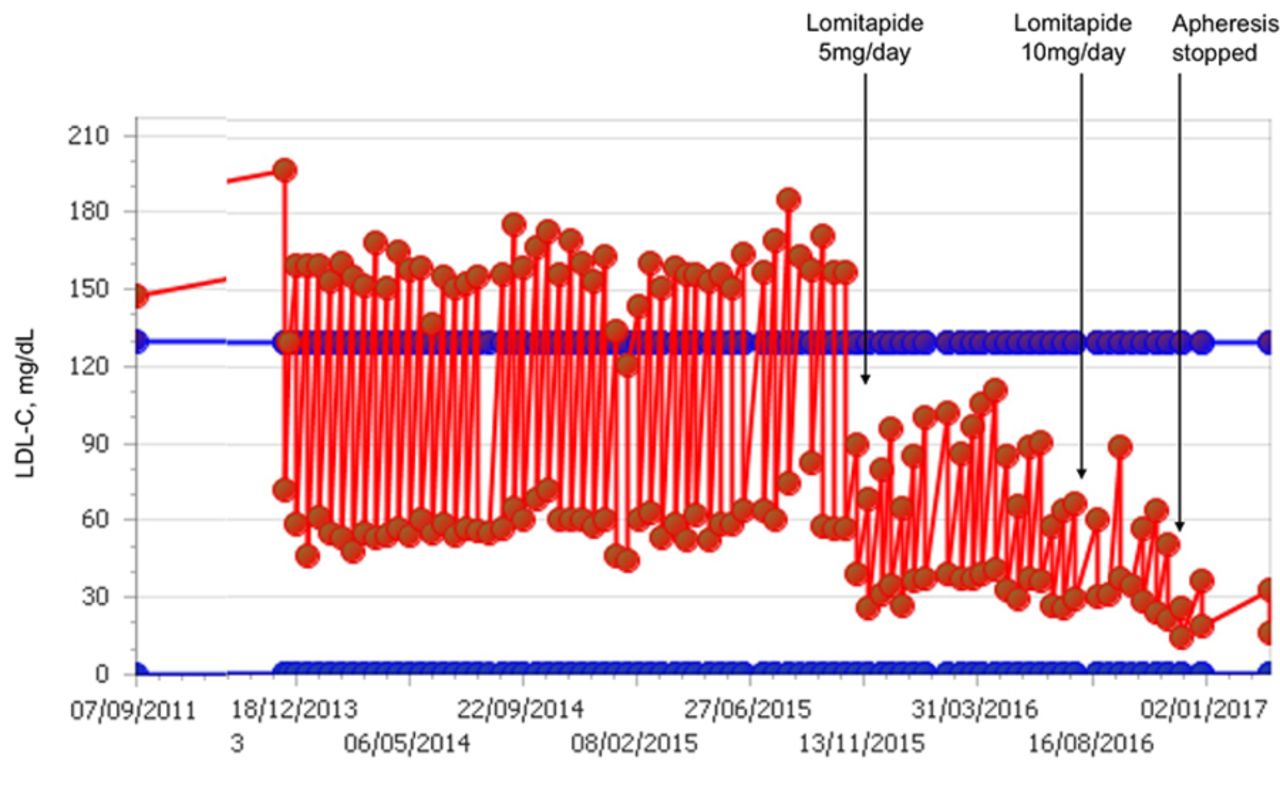
LDL-C values at baseline and during LDL-C apheresis with different doses of lomitapide in Patient 1. LDL-C, low-density lipoprotein cholesterol.
LDL-C pre-apheresis, post-apheresis and inter-apheresis values at baseline, before LDL-C apheresis period and during apheresis period, with different doses of lomitapide in Patient 1
Table 1 shows the LDL-C values in response to lomitapide 5 mg/day and 10 mg/day. Peak and nadir LDL-C levels were immediately lower for lomitapide than before the drug was used. Mean interval LDL-C levels were in the range 25–70 mg/dL (figures 1 and 2, table 1). During this time, no significant increase in transaminases and no alteration in hepatic fibroscan were observed.

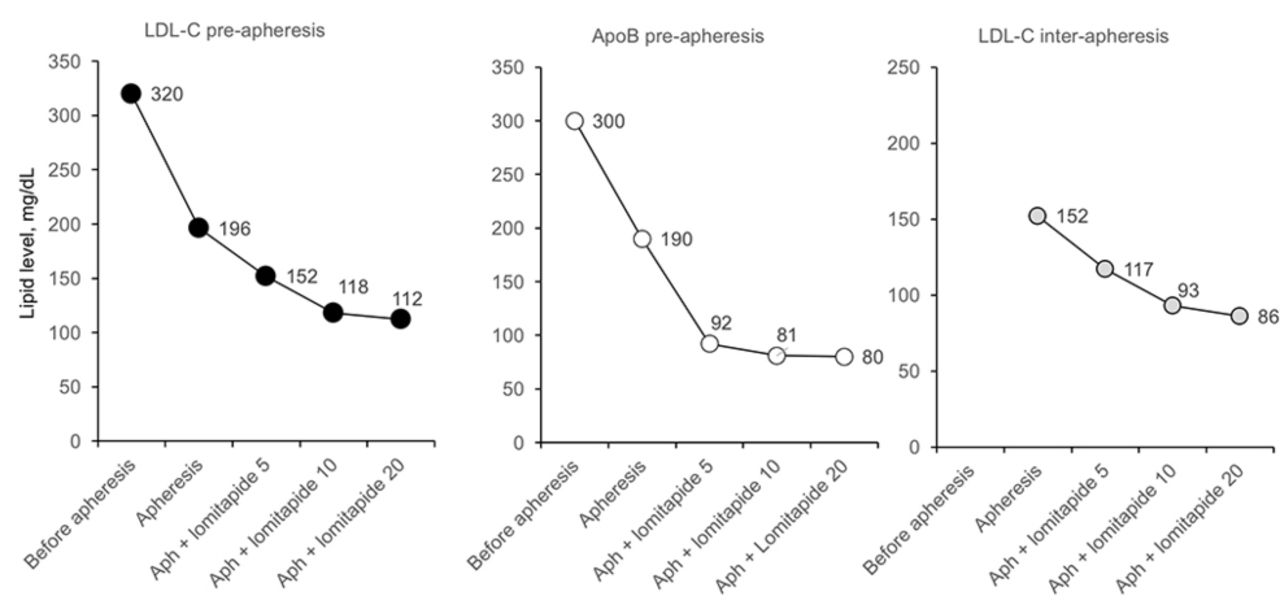
Evolution of LDL-C preapheresis, apoB preapheresis and LDL-C interapheresis values with different doses of lomitapide in patient 1. Aph, apheresis; ApoB, apolipoprotein B; LDL-C, low-density lipoprotein cholesterol; lomitapide doses are mg/day.
Patient 2
At the age of 45 years, the patient changed his lipid-lowering therapy to rosuvastatin 40 mg plus ezetimibe 10 mg and added lomitapide 5 mg/day. Lomitapide was gradually uptitrated to 40 mg/day with progressive decreases in mean interval LDL-C (figures 3 and 4, table 2). At the 40 mg/day dose, he suffered from headache, and the dose was decreased to 20 mg/day with resolution of the symptoms.

LDL-C values at baseline and during LDL-C apheresis with different doses of lomitapide in patient 2. LDL-C, low-density lipoprotein cholesterol.

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Evolution of LDL-C preapheresis, apoB preapheresis and LDL-C interapheresis values with different doses of lomitapide in patient 2. Aph, apheresis; ApoB, apolipoprotein B; LDL-C, low-density lipoprotein cholesterol; lomitapide doses are mg/day.
LDL-C preapheresis, postapheresis and interapheresis values at baseline, before LDL-C apheresis period and during apheresis period, with different doses of lomitapide in patient 2
Outcome and follow-up
Patient 1
Figure 2 shows LDL-C preapheresis, ApoB preapheresis and LDL-C interapheresis values for lomitapide 5–10 mg/day. Due to the good response to lower doses of lomitapide and absence of side effects, the patient was maintained on this regimen.
In 2016, we extended the apheresis interval to one per month, and preapheresis LDL-C levels remained <70 mg/dL. Finally, the patient was able to discontinue apheresis entirely.
Patient 2
At present, the patient has interapheresis LDL-C levels <100 mg/dL, takes lomitapide 20 mg/day and shows no evidence of CV disease. Lomitapide 20 mg/day is well tolerated, and no significant increases in transaminases have been observed. The most recent hepatic fibroscan was normal.
Discussion
In this cases series, we have looked at two patients who our clinical team diagnosed as having HoFH and were therefore candidates for lomitapide therapy. In both cases, we were faced with patients with ASCVD and persistently high LDL-C levels, in spite of maximal tolerable LLT.
In both cases, diagnosis of HoFH was supported by genetic testing. However, we wish to stress that genetic testing is not definitive due to the large number of hitherto unidentified LDLR mutations. In our cases, untreated LDL-C levels were in excess of 500 mg/dL. However, recent studies of patients with genetically confirmed HoFH have demonstrated LDL-C levels (treated and untreated) much lower than historical criteria.16 17 Therefore, the diagnosis of HoFH should not be based on an arbitrary LDL-C threshold; rather, other considerations must be taken into account including physical findings (ie, xanthomas and corneal arcus), family history, premature ASCVD and genetic factors.18
In our cases, the family history of elevated LDL-C combined with clinical signs observed in the patients would have resulted in a diagnosis of HoFH even in the absence of a genetic test. Both patients showed xanthomas at 6 and 8 years, LDL-C values >500 mg/dL and parents with clinical and biological criteria of heterozygous FH. Although the untreated LDL-C values in our patients were very similar, and due to their close relation, the causative mutations were identical, recent evidence has taught us that the number of genes associated with HoFH is more than previously believed, and current estimates place the number of identified LDLR mutations in excess of 1700.4 This drives wide phenotypic variability evidenced by an enormous spectrum of untreated and treated LDL-C levels4 and the variable presence of the characteristic xanthoma and/or corneal arcus.1
Both of our patients exhibited a lack of adequate response to conventional LLT. The lack of effectiveness of standard LLT is not unexpected in patients with a clinical and/or genetic diagnosis of HoFH. Statin therapy, as well as ezetimibe, relies on the upregulation of functional LDL-Rs to exert its effect.19 In HoFH, the LDL-R is either functionally significantly compromised or absent,1 so effective LLT cannot rely on the LDL-R pathway. LA works independently of the LDL-R and is often used to treat HoFH patients. In both our cases, the apheresis procedure was very successful—LDL-C levels dramatically decreased over the course of each session. However, over the interval between sessions, the LDL-C levels rebounded to preapheresis levels. This rebound is consistent with the general sawtooth pattern seen for patients receiving apheresis (see figures 1 and 3).11 20 Rather than looking at the LDL-C nadir to determine if patients remain exposed to lethally high levels of LDL-C, the mean interval measurement is more informative, and in both our cases, patients showed evidence of exposure to continued high levels of LDL-C despite apheresis treatment.
The first of our cases is possibly the most striking. This patient, with renal failure and multiple concomitant medications, some of which are known to interfere with the pharmacokinetics of lomitapide, would not have been a candidate for a clinical trial. Data from this type of patient are important for understanding the effectiveness of drugs in a ‘real-world’ clinical setting.
This patient, although responsive to apheresis, was still exposed to high mean interval LDL-C levels. Lomitapide was started at its lowest dose, and the patient experienced a dramatic reduction in LDL-C values, with no need to further titrate the drug.
In the phase 3 clinical trial of lomitapide, doses of lomitapide were titrated according to a maximum tolerated dose protocol.15 This means that the mean doses in the trial were artificially elevated. In our patient, lomitapide 5–10 mg/day was sufficient to reduce LDL-C levels into the target range suggested by the EAS.1 This remarkable response to low-dose lomitapide allowed the patient to stop apheresis.
In our second patient, lomitapide was titrated up to 40 mg before satisfactory efficacy was achieved, but this is still below the maximum dose allowable in the phase 3 study. Unfortunately, this patient needed to decrease the dose due to headache. However, he still tolerated 20 mg/day well and was able to achieve preapheresis LDL-C <100 mg/dL with an absence of CV episodes. In these two brothers, no side effects were reported (except for the dose-managed headache). Systematic fibroscan of the liver showed no alterations of concern. No elevations of transaminases were observed during the follow-up.
The findings of this small case series are in line with other reports of case series in patients receiving lomitapide to treat HoFH. In a report of seven Italian patients receiving LLT for HoFH, LDL-C reductions of more than 50% were achievable with a range of lomitapide doses from 10 mg/day to 40 mg/day, again, with good levels of tolerability.21
In summary, in these two real-world cases of brothers with HoFH, lomitapide was observed to be an effective adjunct to standard LLT, including apheresis, and was generally well tolerated. Required lomitapide doses appear to be lower in the real world than in the phase 3 clinical trial, and this may assist in the management of AEs.
Learning points
Two brothers with homozygous familial hypercholesterolaemia were treated in our clinic using multimodal lipid-lowering therapy.
Addition of lomitapide to existing therapy resulted in marked reductions in low-density lipoprotein cholesterol (LDL-C) levels.
Doses of lomitapide required to reach LDL-C goals were lower than mean doses in clinical trials.
Acknowledgments
Editorial assistance in the preparation of the manuscript was provided by Nigel Eastmond of Eastmond Medicomm Limited, and was funded by Amryt Pharmaceuticals DAC.
References
Footnotes
Contributors JR: direct responsibility for patient care; wrote, edited and reviewed all drafts; and approved final draft for submission. CA, RG and JFA: direct responsibility for patient care; edited and reviewed all drafts; and approved final draft for submission. Editorial assistance in the preparation of the manuscript was provided by Nigel Eastmond of Eastmond Medicomm Limited, and was funded by Amryt Pharmaceuticals DAC.
Competing interests None declared.
Patient consent Obtained.
Provenance and peer review Not commissioned; externally peer reviewed.