Article Text

Summary
A 65-year-old woman with a 35-year history of limited cutaneous systemic scleroderma was admitted to our hospital complaining of a 3-month history of progressive dyspnoea on exertion. High-resolution CT images of the chest revealed diffuse reticular opacities and traction bronchiectasis predominantly in the bilateral lower lobes of the lung. Specimens obtained during video-assisted thoracic surgery were consistent with fibrocellular non-specific interstitial pneumonia and accompanied by accumulation of lymph follicles within areas of fibrosis. Although the patient received combination therapy with prednisolone and intravenous cyclophosphamide at a dosage of 500 mg/m2 monthly for 5 months, her clinical condition deteriorated gradually. In addition, right heart catheterisation revealed borderline pulmonary arterial hypertension with mean pulmonary artery pressure of 24 mm Hg. Therefore, we initiated a combination therapy of an antifibrotic agent, pirfenidone for 12 months, and the dual endothelin receptor antagonist, macitentan, with prednisolone. As a result, her clinical condition improved dramatically.
- interstitial lung disease
- pulmonary hypertension
- connective tissue disease
- drugs and medicines
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Background
Systemic scleroderma (SSc)-related interstitial pneumonia (IP) and pulmonary artery hypertension (PAH) are leading causes of morbidity and mortality in patients with SSc.
The efficacy and safety of novel antifibrotic agents such as pirfenidone and nintedanib in patients with SSc–IP are currently being evaluated in controlled clinical trials.1
However, with the emergence of evidence-based treatments, including a novel agent such as macitentan, and the focus on early detection of SSc–PAH, there has been considerable improvement in patient survival. Moreover, patients with SSc may develop borderline pulmonary artery pressure (PAP), which may represent the early stage of PAH.
Herein, we describe a case of long-term efficacy and safety of combination therapy of macitentan and pirfenidone in a patient with SSc–IP associated with borderline PAH.
Case presentation
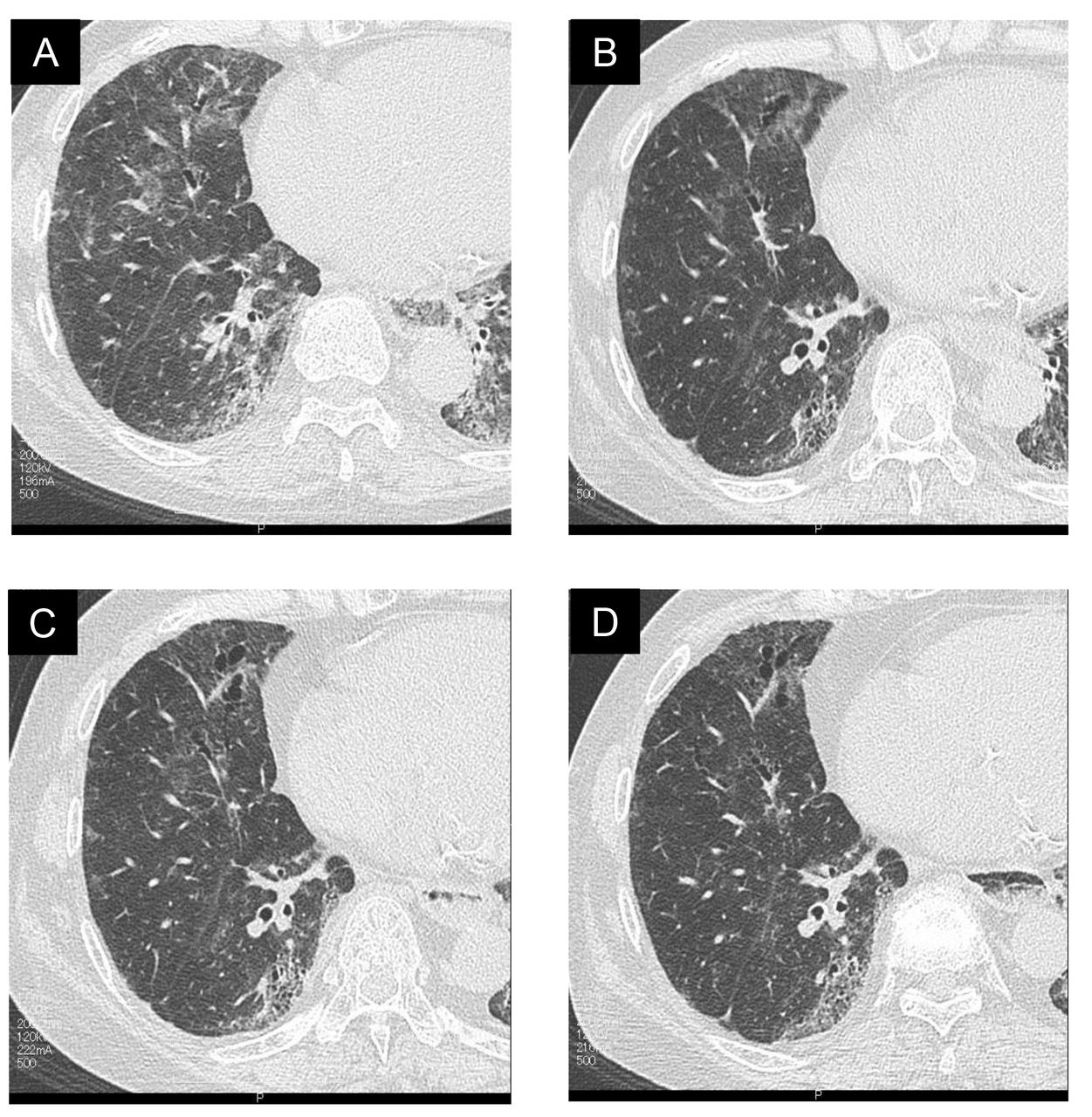
A 65-year-old woman with a 35-year history of limited cutaneous SSc was admitted to our hospital for progressive dyspnoea on exertion with onset at 3 months before admission. Physical examination revealed scleroderma in both arms and legs, with puffy fingers, Raynaud phenomenon and ankyloglossia. Lung auscultation revealed fine crackles in both lung bases. Laboratory test data from serum showed high levels of Krebs von den Lungen-6 (1223 U/mL), surfactant protein D (128 ng/mL), brain natriuretic peptide (BNP, 39.5 U/mL), antinuclear antibody titre (320-fold) and antiribonucleoprotein antibody titre (64-fold). Arterial blood gas analysis showed a pH of 7.41, arterial carbon dioxide tension of 41.5 mm Hg and arterial oxygen tension of 80.6 mm Hg at room air. The pulmonary function test revealed restrictive impairment (forced vital capacity (FVC) of 1.81 L and 77.7% of predicted) with decreased diffusion capacity for carbon monoxide (8.78 mL/min/mm Hg and 49.4% of predicted). Diffuse reticular and widespread ground-glass opacity (GGO) shadows and interlobular septal thickening in both middle and lower lobes, without honeycombing, were evident on chest high-resolution CT (HRCT) scan (figure 1A and B). The lung biopsy specimens of the left lung segments, S8 and S10, obtained by video-assisted thoracic surgery (VATS), revealed fibrocellular non-specific IP (NSIP) accompanied by accumulation of lymph follicle formation (figure 2A and B). Additionally, small pulmonary arteries with intimal fibrosis and medial hypertrophy in fibrotic lesions were widespread, resulting in marked luminal narrowing (figure 2C and D). Consequently, the patient was diagnosed with fibrocellular NSIP associated with SSc. Abnormalities of chest HRCT images, especially GGO, were initially ameliorated with the use of methylprednisolone pulse therapy (figure 3A and B). Although she received a combination therapy of prednisolone and intravenous cyclophosphamide (CY) at a dose of 500 mg/m2 monthly for 5 months, her clinical symptoms and pulmonary function deteriorated gradually as deduced from pulmonary function tests and chest HRCT (figure 3C). Right heart catheterisation demonstrated borderline PAH with a mean PAP (mPAP) of 24 mm Hg, mean pulmonary capillary wedge pressure of 9 mm Hg, pulmonary vascular resistance of 185.5 dyne/s/cm5 and cardiac output of 6.47 L/min. The serum level of BNP increased from 39.5 U/mL to 67.4 U/mL. Moreover, the FVC (%) decreased from 77.7% to 65.8%.

Diffuse reticular and widespread GGO shadows and interlobular septal thickening in both middle and lower lobes, without honeycombing, were evident on the chest CT scan. (A) Transverse section on chest HRCT (left lower lobe). (B) Coronal images of chest CT. GGO, ground-glass opacity; HRCT, high-resolution CT.

The lung biopsy specimens of the left S8 and S10 obtained by VATS show fibrocellular NSIP accompanied by accumulation of lymph follicle formation. (A) Elastic van Gieson stain, scale bar=1 mm. (B) H&E stain, scale bar=200 µm. There are widespread small pulmonary arteries with intimal fibrosis and medial hypertrophy in fibrotic lesions, resulting in marked luminal narrowing. (C) H&E stain, scale bar=100 µm. (D) Elastic van Gieson stain, scale bar=100 µm. NSIP, non-specific interstitial pneumonia; VATS, video-assisted thoracic surgery.

{kind=link}
{kind=link}
{kind=link}
(A) Chest CT scan shows diffuse reticulation and GGO and interlobular septal thickening in both middle and lower lobes at initial visit. (B) Abnormalities of chest CT images, especially GGO, are initially ameliorated with the use of methylprednisolone pulse therapy. (C) After 12 months from initial treatments, patchy GGO on chest CT deteriorates gradually. (D) After administration of pirfenidone, patchy GGO on chest CT is associated with a trend towards improvement. GGO, ground-glass opacity.
Treatment
We initiated a combination therapy of pirfenidone and macitentan, with prednisolone. After 6 months of receiving this combination therapy, her clinical condition gradually improved, and mPAP decreased from 24 mm Hg to 15 mm Hg. The serum level of BNP also decreased to 30.1 U/mL. In addition, FVC increased to 74.3%. The 6 min walking distance (6MWD) and patchy GGO on chest HRCT scans were associated with a trend towards improvement (figure 3D). After receiving this combination therapy for 12 months, her clinical condition was stable with an mPAP of 17 mm Hg (table 1). She did not experience any serious adverse effects.
Hemodynamic, 6 min walking test, and pulmonary function parameters before and after combination therapy of pirfenidone and macitentan.
Discussion
To the best of our knowledge, this is the first report of a case where the combination therapy of pirfenidone and macitentan demonstrated efficacy in a patient with limited cutaneous SSc, fibrocellular NSIP and borderline PAH, which may translate into a favourable prognosis.
However, our results suggest that an improvement in pulmonary function with pirfenidone treatment does not necessarily correlate with an improvement in oxygenation.
SSc is a systemic idiopathic autoimmune disease characterised by exaggerated extracellular matrix deposition in skin and various internal organs, severe fibroproliferative vasculopathy and cellular and humoral immune response abnormalities. SSc–IP and SSc–PAH are both leading causes of morbidity and mortality in patients with SSc. In particular, patients with an mPAP between 21 and 24 mm Hg are considered to represent so-called borderline PAH. Additionally, their condition is thought to represent the early stages of pulmonary arterial vasculopathy, which is also an intermediate stage between normal PAP and the manifestation of PAH. In fact, the rate of progression to PAH in patients with SSc and associated borderline PAH was 19% after 3 years and 27% after 5 years.2 Kovacs et al3 recently reported that the borderline elevation of PAP was associated with cardiac and pulmonary comorbidities, decreased exercise capacity and a poor prognosis. In the present report, the patient only experienced improvement in mPAP and oxygenation after macitentan administration. Therefore, we believe that early interventions, such as administration of dual endothelin receptor antagonists, may play an important role in improving the treatment and outcome of patients with SSc and borderline PAH.
According to the LOTUSS trial,1 pirfenidone had an acceptable tolerability profile in patients with SSc–IP. Moreover, preliminary studies evaluating the safety and efficacy of pirfenidone and nintedanib in patients with SSc–IP are currently underway. Two recent case series reported that patients with SSc–IP treated with pirfenidone showed improvement of dyspnoea, increased vital capacity and less GGOs.4 5 More recently, Tashkin et al6 reported that efficacy of treatment with mycophenolate mofetil for SSc-interstitial lung disease (ILD) was similar to that of CY therapy and better tolerated. In addition, patients with IP related to connective tissue diseases are not usually needed to biopsy under VATS in clinical setting. However, we suppose that this attempt is useful to select whether antifibrotic agents or anti-inflammatory agents such as corticosteroid or immunosuppressants should be introduced. Indeed, this patient was diagnosed with fibrocellular NSIP histopathologically. The long-term efficacy of combination therapy of pirfenidone and macitentan in patients with SSc–IP associated with secondary PAH has not yet been proven. However, improvements in the modified Medical Research Council dyspnoea scale, 6MWD and pulmonary haemodynamic parameters in the present case indicated an increased exercise capacity. Nevertheless, the patient consistently presented the values of lowest peripheral capillary oxygen saturation (SpO2) during the 6 min walking test (6MWT). We presume that an improvement in pulmonary function with pirfenidone treatment does not always correlate with the improvement in oxygenation because, in previous clinical trials of pirfenidone, no statistically significant differences have been detected in changes in SpO2 during the 6MWT, which remained low. Our patient remained stable throughout the 1-year follow-up period. Of note, our patient did not die or experience serious drug-related adverse events during treatment.
In conclusion, long-term (12-month) combination therapy of pirfenidone and macitentan can provide a clinical and radiological improvement for patients with SSc–IP and borderline PAH when conventional treatments, such as prednisolone and/or CY, are ineffective.
Learning points
The long-term (12-month) combination therapy of pirfenidone and macitentan may provide a clinical and radiological improvement for patients with systemic scleroderma-related interstitial pneumonia and borderline pulmonary artery hypertension when conventional treatments, such as prednisolone and/or cyclophosphamide, are ineffective.
Footnotes
Contributors KSu and SH: study design, data analysis, manuscript preparation and guarantor of the paper. KSu, TK and KSh: data collection and data analysis. KSu, KSh and SH: manuscript preparation and review. All authors had full access to all of the data in the study and can take responsibility for the integrity of the data and the accuracy of the data analysis.
Funding The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests None declared.
Patient consent Obtained.
Provenance and peer review Not commissioned; externally peer reviewed.