Article Text

Statistics from Altmetric.com
Description
A 15-year-old boy presented with a history of absence of fingernails and toenails since birth. He was the first born of a non-consanguineous marriage, delivered vaginally at term with normal birth weight. His mother admitted to excessive consumption of opioid analgesics (ethyl morphine derivatives) during all three trimesters of her first pregnancy, which she discontinued subsequently. He was otherwise healthy, with age-appropriate growth, development and intelligence. His parents and two younger siblings were unaffected.
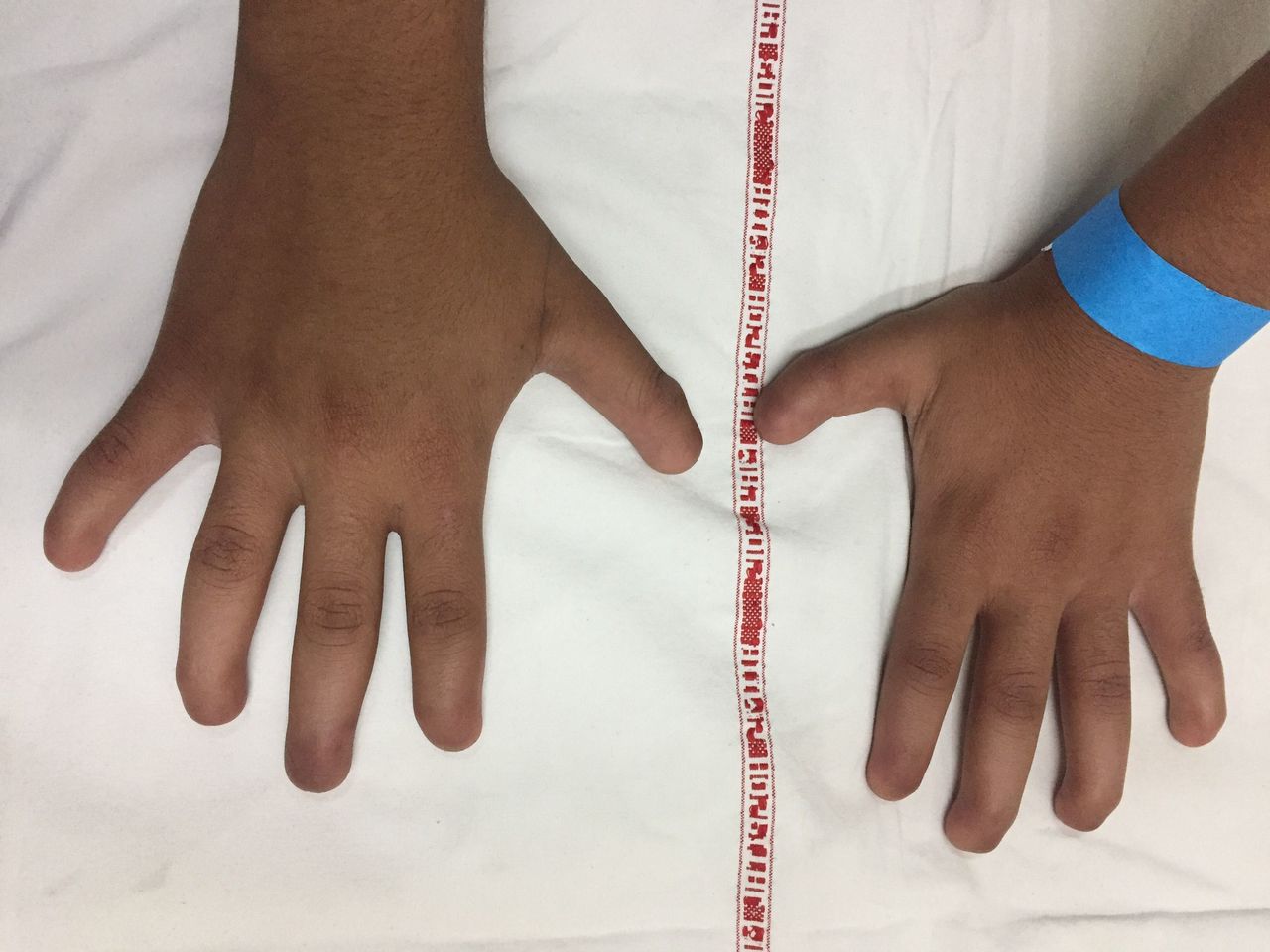
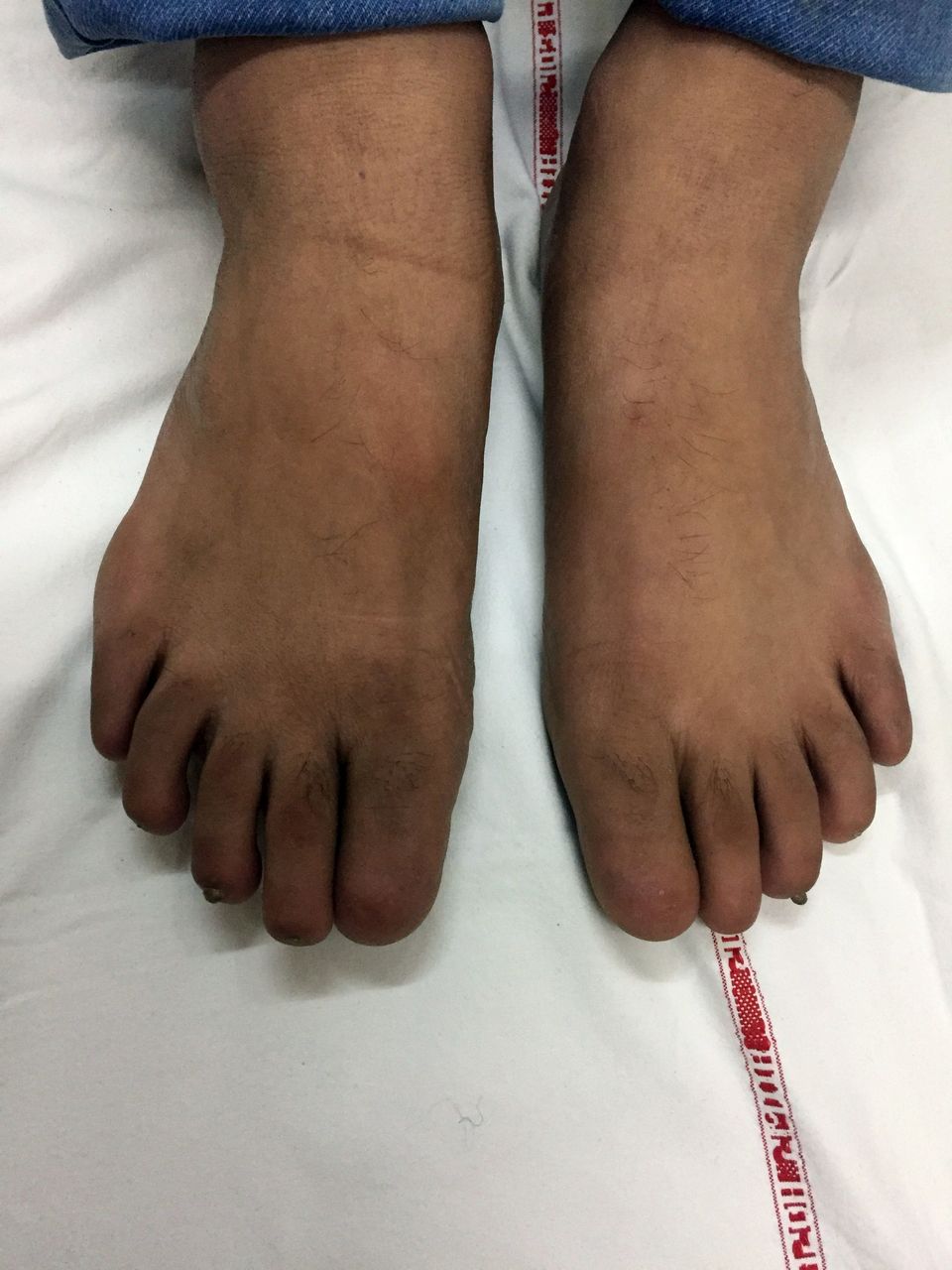
Examination revealed complete absence of all 20 fingernails and toenails (figure 1). Tips of digits were soft, with absence of nail folds, but small keratinised nail plates were present in middle toes bilaterally (figure 2). Skin, teeth and hair were normal. Rest of the general physical and systemic examination was unremarkable with no dysmorphic features. Radiography of hands revealed hypoplasia of terminal phalanges involving all digits, suggestive of acroosteolysis (figure 3). Rest of the skeletal survey was normal. Visual acuity and pure tone audiometry were normal. Clinically, the patient was diagnosed with congenital anonychia totalis possibly secondary to teratogenic effect of morphine.

Photograph of hands showing absence of fingernails bilaterally.

Photograph of feet showing absence of toenails, with small keratinised nail plates present in bilateral middle toes.

{kind=link}
{kind=link}
{kind=link}
Radiograph of hands showing hypoplastic terminal phalanges in all digits suggestive of acroosteolysis.
Anonychia (absence of nails of fingers and toes) can be acquired or congenital. A search in English language up to July 2017 identified only 21 reported cases of the congenital anonychia. It most often occurs as a part of syndromes with serious limb and skeletal defects such as nail–patella syndrome; ectodermal dysplasia; anonychia–lymphoedema or DOOR (deafness, onychoosteodystrophy, mental retardation)syndrome.1 Isolated, non-syndromic congenital anonychia is an extremely rare condition, usually following an autosomal recessive inheritance and has been mapped to defects in chromosome 20p13. Analysis of R-spondin 4 (RSPO4) gene (belonging to R-spondin family of proteins) on this locus, in affected individuals, has revealed frameshift, splice site and missense mutations in exon 2.2 RSPO4 gene product is implicated in launch of Wnt/beta-catenin signalling pathway, playing a pivotal role in mesenchymal–epithelial interactions and embryonic development.
Nail plate originates from germinative epithelium of nail matrix and grows continuously, from 15 weeks of embryonic life until death. Maternal consumption of drugs during early pregnancy can significantly alter nail development, and defects have been reported with use of alcohol, carbamazepine, phenytoin, warfarin, morphine and trimethadione.3 This could be the possible aetiology in our patient, though genomic sequencing is required to confirm absence of known causal mutations. Growth of phalanges is responsible for characteristics such as outline and curvature of the nails; hence, abnormality of the two together is a common occurrence. This could justify the presence of acroosteolysis seen in our patient.
Anonychia due to acquired causes includes trauma, psoriasis, severe lichen planus and Stevens-Johnson syndrome and can potentially reverse with treatment of the underlying pathology.
Our patient had congenital anonychia as an isolated and asymptomatic condition which did not affect his day-to-day activities or cause any inconvenience. He was reassured, and prosthetic acrylic nails were provided for cosmesis. In such cases, it is crucial to screen for skeletal, neurological, visual, auditory and dermatological coassociations. Patients with suspected familial inheritance or proven RSPO4 gene mutations should avoid close relatives for a mate.
Patient’s perspective
Since the time I started going to school, I realised I was different, and the unusual appearance of my hands and feet used to be a subject of mockery. As I grew, I became used to this appearance; however, the thought of having a serious underlying illness used to haunt me. It affected my work and studies and turned me into a recluse. Now, after having detailed discussions with my doctors, I have understood about my disease and its rarity. I am extremely relieved on learning that it has no serious long-term implications, and I am as healthy as any of my peers. Also, my new prosthetic nails have improved the aesthetic look and made me more confident. I am glad that people can know about this condition through my case, so that they can seek appropriate medical advice for themselves and their family members to detect any significant underlying illness.
Learning points
Isolated, non-syndromic congenital anonychia is a rare entity with autosomal recessive inheritance.
Mutations in R-spondin 4 gene involving Wnt/beta-catenin pathway have been implicated.
Maternal use of alcohol, antiepileptics and morphine in early pregnancy can cause anonychia and hyponychia.
Screening for associated skeletal, dermatological and other systemic defects is essential.
Footnotes
Contributors AM and PJ were the primary physicians of the patient. AM reviewed the literature and prepared the manuscript. PJ edited and finalised the manuscript.
Competing interests None declared.
Patient consent Obtained.
Provenance and peer review Not commissioned; externally peer reviewed.