Article Text

Summary
Sarcoidosis and lymphoma are generally thought of as being two mutually exclusive diseases that need to be considered in the differential diagnosis of patients with hilar/mediastianal lymphadenopathy. However, there are rare patients in whom both of these diseases coexist. These patients constitute a diagnostic challenge because their presentation (ie, clinical symptoms, imaging abnormalities and even pathology) may all be atypical when each individual disease is considered separately. In this report, we describe a patient who presented with such atypical features and was eventually diagnosed as having both sarcoidosis and a B-cell lymphoma with features of splenic marginal zone lymphoma (SMZL) simultaneously. To our knowledge, this is only the second reported case of SMZL and sarcoidosis in the same patient.
- immunology
- cancer
- respiratory system
- cancer intervention
- pathology
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Background
Sarcoidosis is a chronic multisystem granulomatous disease of unknown aetiology. The association of sarcoidosis and lymphoproliferative disorder (LPD) has been previously described in the literature with less than 79 cases reported.1 Occurrence of LPD in patients with chronic active sarcoidosis is becoming increasingly recognised as it creates a diagnostic challenge. Both diseases have many common presentations, including constitutional symptoms, peripheral and mediastinal lymphadenopathy, haematological abnormalities, hypercalcaemia and similar organ involvement such as lung, liver and spleen. It has been speculated that immunological abnormalities associated with sarcoidosis may predispose patients to LPD.2 In this case report, we present splenic marginal zone lymphoma (SMZL) that surfaced in a patient with sarcoidosis.
Case presentation
A previously healthy 43-year-old Caucasian woman presented with a 2-month history of productive cough, fever, dyspnoea, fatigue and occasional night sweats. She received several courses of oral antibiotics without any improvement in her symptoms. Physical examination revealed diffuse bilateral fine crackles, palpable left axially lymph node and splenomegaly. Complete blood count showed normal white cells with normal differential and mild anaemia (haemoglobin concentration 11g/dL). Pulmonary function tests showed lung restriction (forced vital capacity 51%, total lung capacity 52%) with normal diffusing capacity (carbon monoxide transfer factor 90%). Oxygen saturation at rest and on room air was 95%, dropping to 87% at the end of 6-min walk test during which she covered a distance of 445 m.
Investigations
CT of the chest (figure 1) showed extensive lymphadenopathy throughout the mediastinum, hilar regions and in both axillae; in the lung parenchyma there were multiple areas of consolidation scattered diffusely, bilaterally, worse at the lung bases. The radiographical appearance was most consistent with neoplastic disease (such as lymphoma or bronchoalveolar cell carcinoma) or inflammatory disease such as cryptogenic organising pneumonia (COP).

Representative CT cuts of the thorax. (A) Contrast-enhanced cut demonstrating a 33×20 mm heterogenously enhancing subcarinal (white arrowheads) and bilateral 16 mm right and 11 mm left interlobar nodes (white arrows). (B) Lung window cut demonstrating bilateral patchy consolidation with basal predominance; the air space disease is a mixture of more nodular foci and larger more confluent areas of consolidation (*).
Bronchoscopy and bronchoalveolar lavage were carried out. Cytology, tuberculosis smear, bacterial and fungal cultures were non-diagnostic. No transbronchial biopsy was performed due to elevated International Normalised Ratio (INR). Left axillary lymph node biopsy showed necrosis, to an extent that precluded determination of the aetiology, although the fact the node was necrotic is more in keeping with possible malignancy rather than a ‘benign’ process. There was no evidence of infection based on special stains and cultures. Presumptive diagnosis of COP was made. The patient was started on a tapering course of prednisone with rapid symptomatic improvement. Repeat CT scan of the thorax showed improvement in the alveolar opacities, but persistence of mediastinal and axillary lymphadenopathy. Concomitant with tapering and eventual discontinuation of prednisone, the patient again started experiencing constitutional symptoms and also developed Bell’s palsy. CT scan of the abdomen showed that the spleen was enlarged, measuring 23 cm.
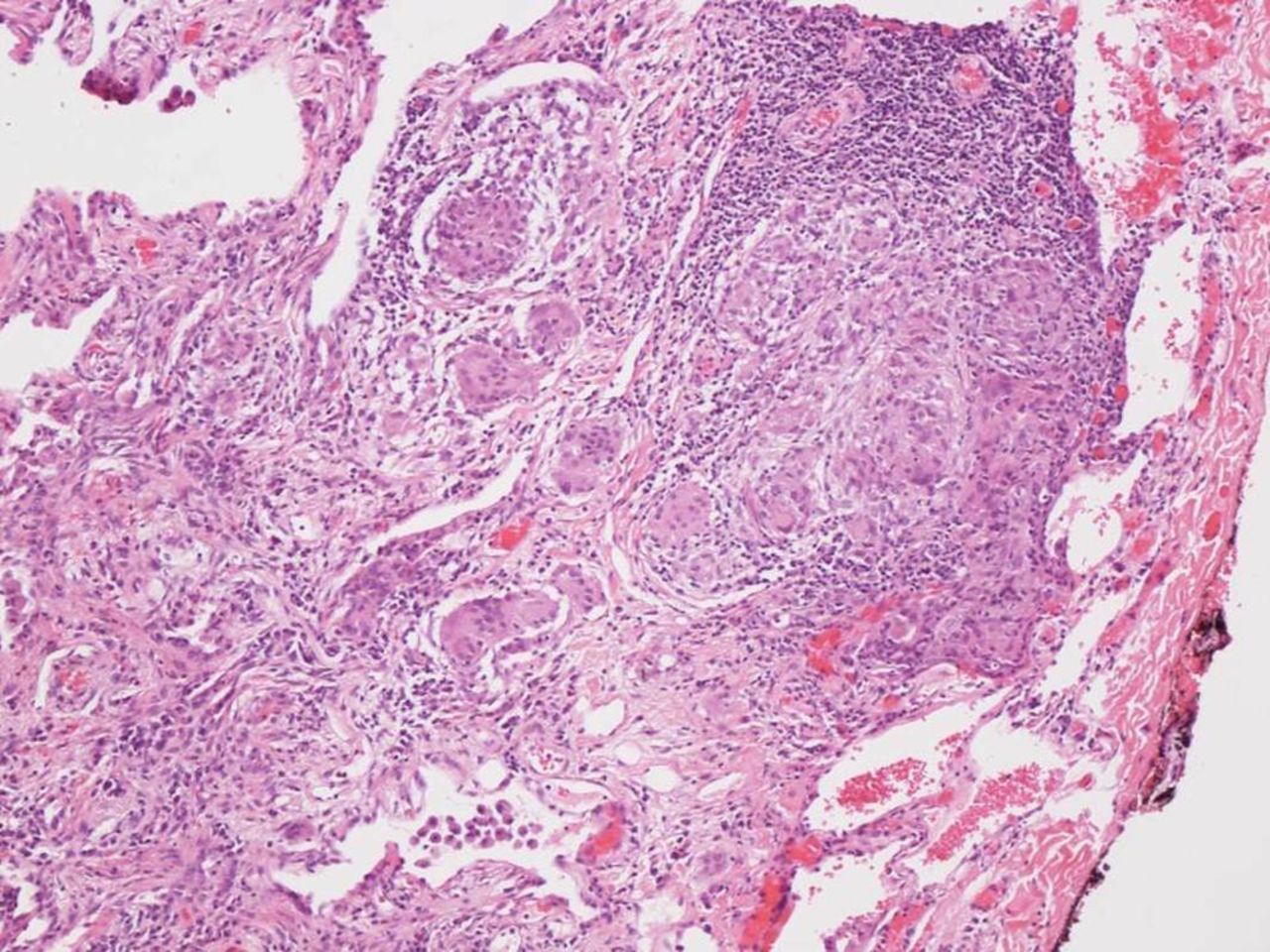
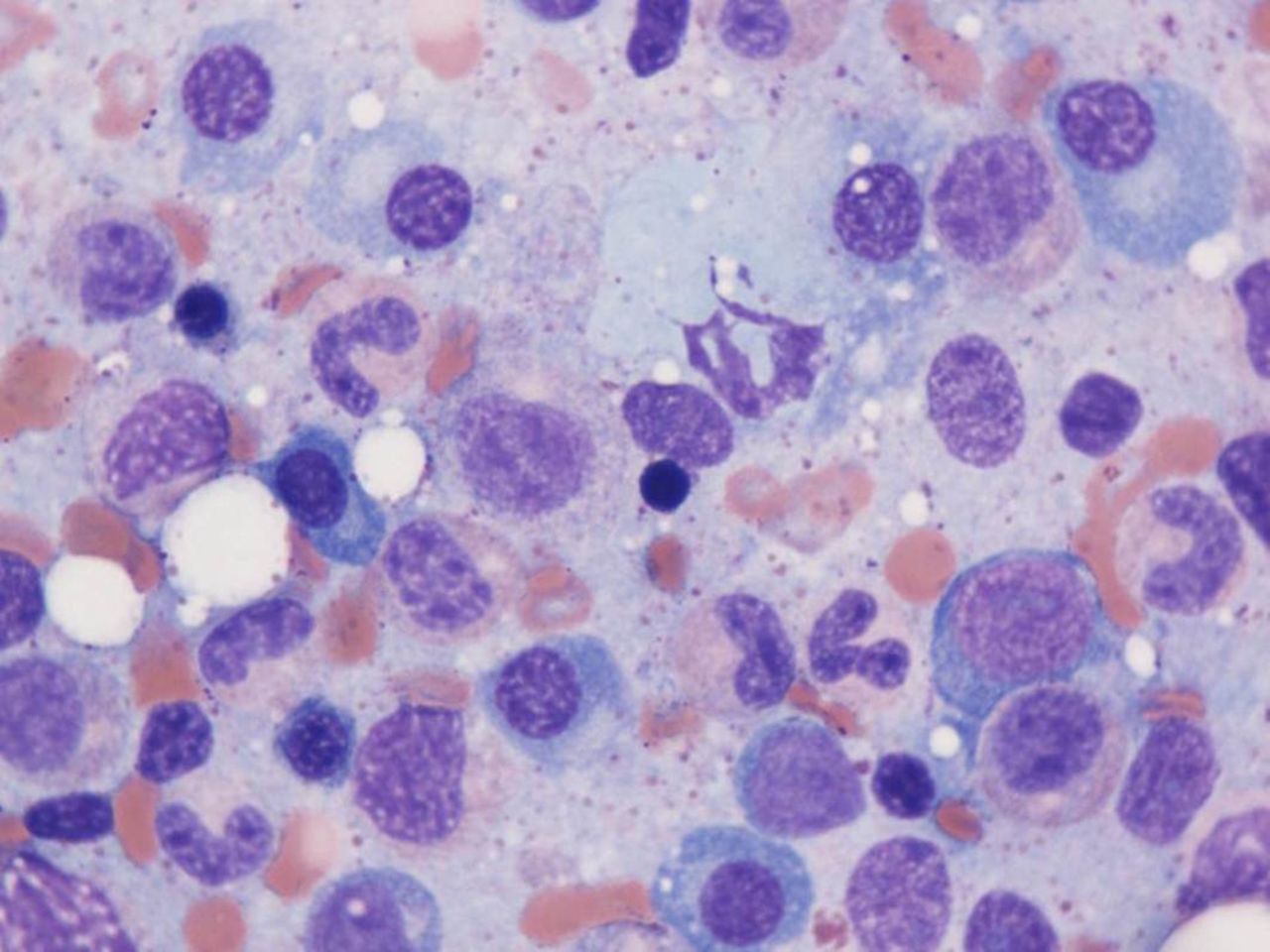
Mediastinoscopy and surgical lung biopsy were carried out. There was no evidence of infection in any of the specimens. Histopathological evaluation of wedge biopsies taken from the right upper, middle and lower lobes revealed multifocal well-formed granulomata that were mainly non-necrotising (figure 2). No granulomata were detected within the lymph nodes, but a population of plasma cells showing lambda light chain restriction by immunohistochemistry was present within the nodal sinuses. Laboratory investigations at this time revealed elevated serum ACE levels and hypercalcaemia—strongly suggestive of sarcoidosis, and treatment with prednisone was reinstituted. At this stage, serum protein electrophoresis, immunofixation and free light chains were also measured, revealing a monoclonal protein consisting of free lambda light chains at a level of 9870 mg/L. Bone marrow aspirate (figure 3) and biopsy showed a population of mature-appearing plasma cells showing evidence of lambda restriction by immunohistochemistry, accounting for approximately 10%–15% of the marrow cellularity. The plasma cells did not display aberrant expression of CD56, CD20, CD117 or cyclin D1 based on immunohistochemical analysis. There was no significant infiltration by lymphocytes and no monotypic B-lymphocyte population was detected by flow cytometry. A clinicopathological diagnosis of myeloma was entertained; however, hypercalcaemia as well as Bell’s palsy promptly resolved with the institution of prednisone and there was no evidence of end-organ dysfunction. Because of the side effects of prednisone, methotrexate was also added and prednisone was gradually discontinued.

Lung, 100×, H&E: Lung parenchyma with well-formed non-caseating granulomata and conspicuous multinucleated giant cells.

Bone marrow aspirate, 1000× oil, Wright-Giemsa: The bone marrow aspirate shows increased numbers of plasma cells. Immunohistochemical studies performed on the bone marrow core biopsy (not shown) showed approximately 10%–15% plasma cells with cytoplasmic light chain restriction.
Outcome and follow-up
However, over the following year, despite being on methotrexate, the patient continued to have recurrent constitutional symptoms, GI symptoms, a parotid cyst, anaemia, peripheral lymphadenopathy and rapidly progressive splenomegaly. Biopsies of the neck nodes, small bowel and colon were all non-diagnostic.
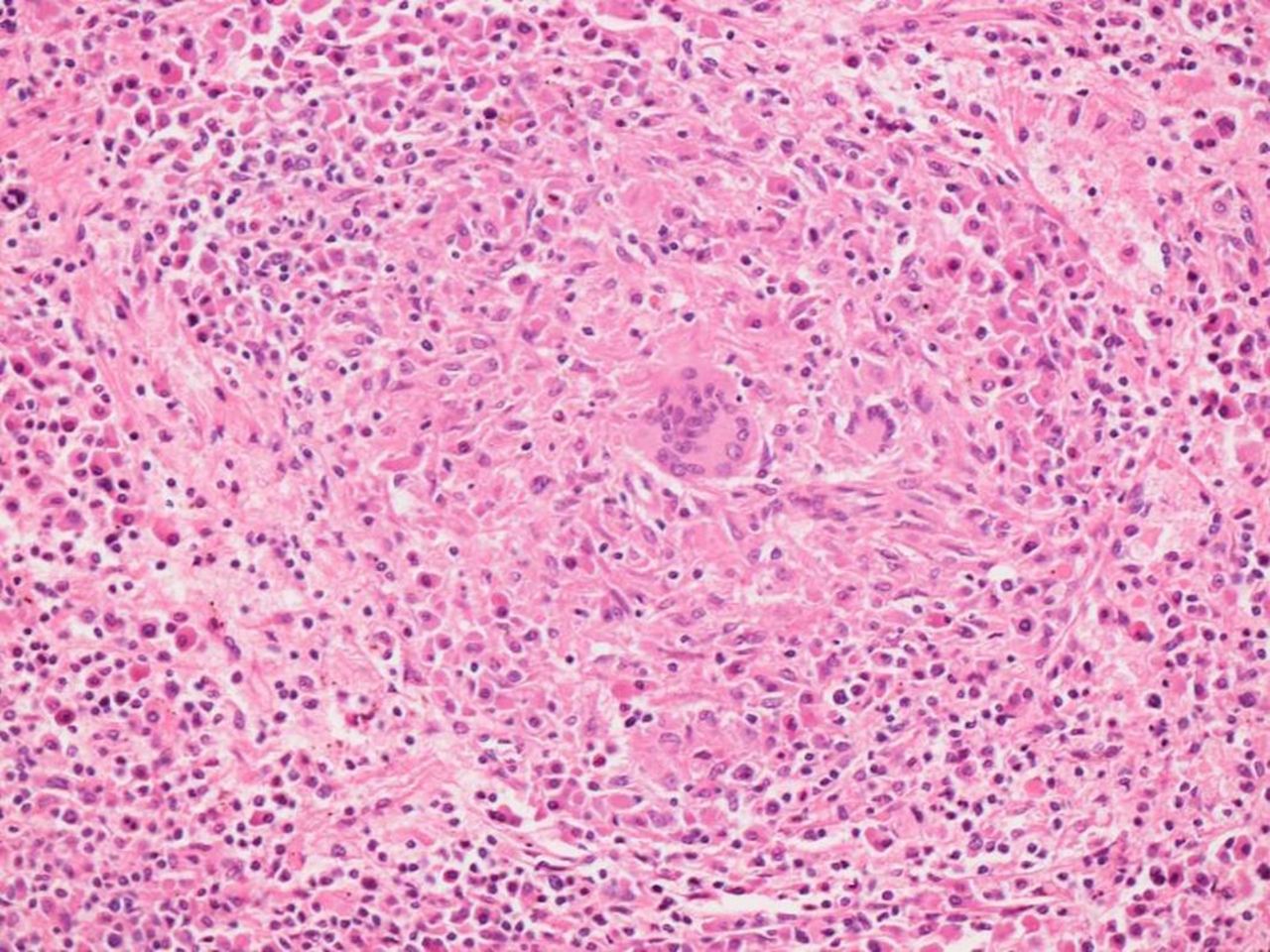
The patient developed progressive left upper quadrant abdominal pain and massive splenomegaly. Splenectomy was carried out (figure 4). Prior to splenectomy white cell count was 3.66 x 109/L with neutrophilic predominance (neutrophil count 3.36, lymphocytes 0.16, basophils 0.10). There was no suggestion of leukaemia. Histopathological assessment revealed a low-grade B-cell lymphoma consistent with marginal zone lymphoma with extensive plasmacytic differentiation reflected by the presence of component of lambda restricted plasma cells involving the spleen and perisplenic lymph nodes (figures 5 and 6). The splenic parenchyma additionally contained multifocal non-necrotising granulomata resembling those noted in the previous lung wedge resection (figure 7). The clinical and pathological findings were in keeping with both marginal zone lymphoma and sarcoidosis. Although the possibility of two distinct clonal processes, plasma cell myeloma in the bone marrow and marginal zone lymphoma in the spleen and perisplenic lymph nodes was entertained, the morphological and immunophenotypical features, including prominent plasmacytic differentiation with lambda restriction at all involved sites and absence of any aberrant markers associated with myeloma in the marrow were considered to be more closely aligned with a unifying diagnosis of marginal zone lymphoma with plasmacytic differentiation. The patient became asymptotic after the splenectomy, but was continued on methotrexate for additional 8 months, thus receiving a total course of prednisone and methotrexate for a total of 13 months. At the most recent follow-up, 7 months after being off these medications, she continued to be asymptomatic with normal serum calcium and serum ACE levels.
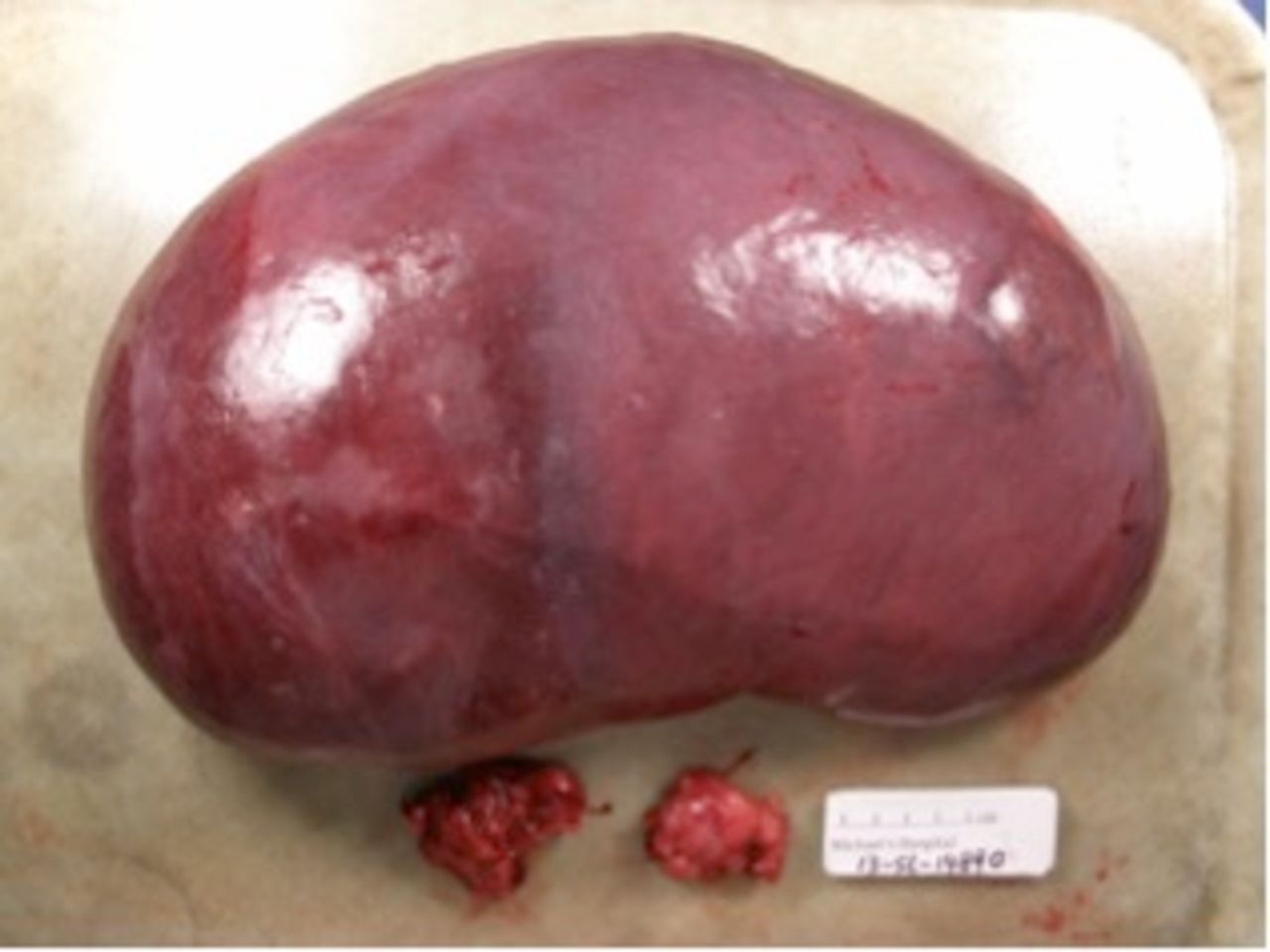
Gross image of intact spleen showing marked splenomegaly (3100 g, 30 cm in maximum dimension) with two enlarged perihilar lymph nodes below.

Splenic hiliar lymph node 100×, H&E: Marginal zone lymphoma associated with effacement of the normal lymph node architecture by a nodular infiltrate of small lymphocytes and plasma cells associated with disruption of follicular dendritic cell meshworks as noted by CD21 staining (inset, immunohistochemistry, 100×).

Spleen 400×, H&E: Marginal zone lymphoma with plasmacytic differentiation involving splenic parenchyma characterised by dense sheet-like proliferation of mature plasma cells with cytoplasmic lambda light chain expression (inset, lambda immunohistochemistry, 400×). Only rare plasma cells labelled with kappa (not shown).

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Spleen 100×, H&E: spleen with non-caseating granulomata.
Discussion
Coexistence of sarcoidosis and lymphoma has been reported previously. In fact, patients with sarcoidosis are up to 11 times more likely to develop lymphoma.2 However, our case is unique in several respects.
As far as the sarcoidosis is concerned, there were a number of unusual features. First, her chest examination showed fine crackles, which is distinctly unusual in sarcoidosis. Second, distribution of lymphadenopathy and parenchymal abnormalities on the imaging were not typical for sarcoidosis. Third, despite the fact that the diagnostic yield of lymph node biopsies, particularly when the lymph nodes are pathologically enlarged, is over 80%—none of this patient’s lymph nodes (axillary and mediastinal) showed granulomatous inflammation; in fact none of the other multiple organ biopsies, except the lung and spleen, showed classic granulomas.
There are also several unusual aspects concerning the lymphoma. First, it was very difficult to establish the diagnosis, despite repeated biopsies of multiple anatomical sites; it was only the splenectomy which allowed us to arrive at a diagnosis that would best explain the clinical picture. Second, the finding of plasmacytosis with lambda restriction in the bone marrow in the setting of elevated monoclonal serum free light chains simulated a plasma cell neoplasm, but the perisplenic lymph nodes showed features typical of marginal zone lymphoma with marked plasmacytic differentiation, suggesting that the marrow findings reflect involvement by the same process. Patients with sarcoidosis–lymphoma syndrome can create a challenging diagnostic dilemma. Granulomatous inflammation alone is not diagnostic of sarcoidosis and may occur in malignancies and infections, similar to our patient. In retrospect, however, there were several features which are common in sarcoidosis but not in lymphoma, such as development of Bell’s palsy and concomitant elevation in serum calcium and serum ACE levels.
The other unusual aspect of this case was the occurrence of SMZL, which is an uncommon type of B-cell lymphoma with an indolent course, which typically affects the spleen and bone marrow. Peripheral lymphadenopathy is extremely rare, accounting for less than 1% of all lymphomas and typically occurs in adults over 50 years of age. SMZL almost always involves the bone marrow with several studies showing 100% incidence rate.3 Marginal zone lymphoma frequently displays plasmacytic differentiation which can occasionally be extensive and may yield pathological findings that are indistinguishable from a plasma cell neoplasm. The hallmark presentation of SMZL is moderate-to-massive symptomatic splenomegaly, which is rarely seen in sarcoidosis.
It is estimated that 7% of patients with sarcoidosis have splenic involvement, which is commonly asymptomatic.4 A study of 79 patients with sarcoidosis–lymphoma syndrome suggested that bilateral hilar lymphadenopathy and presence of lung disease is highly suggestive of sarcoidosis versus lymphoma. On the other hand, presence of splenomegaly and bone marrow involvement was a common feature of lymphoma patients. Mediastinal and peripheral lymphadenopathy as well as constitutional symptoms occur in both diseases and are not clinically helpful to distinguish between the two.
Perhaps the most interesting aspects of this case are the temporal evolution of these diseases and the remarkable disease control of both sarcoidosis and lymphoma that resulted from splenectomy. It is not possible to determine which entity was the primary driver in this case. Was the sarcoidosis driving the lymphoma or vice versa? Although we cannot answer these questions definitively, the patient’s response to treatment gives us some clues. Our patient responded well to prednisone, as expected in sarcoidosis, but SMZL was likely only temporarily suppressed by this treatment. We hypothesise that the persistence and ultimately the progression of lymphoma after prednisone was discontinued, may have induced recurrence of sarcoidosis as well. In contrast, once the SMZL was effectively treated by splenectomy, the sarcoidosis responded well to treatment as manifested by the fact that there was no further recurrence after 7 months post-treatment. We suspect that the two diseases were synergistic and that each needed to be fully and appropriately treated before either could resolve.
Learning points
Hilar and mediastianal lymphadenopathy are common in sarcoidosis and lymphoma, and there are rare patients in whom both of these diseases coexist, particularly when splenomegaly becomes a dominant feature.
Unexplained recurrent peripheral lymphadenopathy and worsening splenomegaly in patient with sarcoidosis must raise suspicion of concurrent lymphoproliferative disease.
In such patients splenectomy may be necessary to establish a diagnosis.
Precise diagnosis, including repeated lymph node biopsies, is essential in such case to avoid inappropriate treatment (eg, prednisone).
Footnotes
Contributors AO is primary author of the paper. The manuscript was edited by HG and LH. VH is the main supervising attending on the case.
Competing interests None declared.
Patient consent Obtained.
Provenance and peer review Not commissioned; externally peer reviewed.