Article Text

Summary
We utilise a clinical case to highlight why exclusion of voltage-gated potassium channel (VGKC)-complex autoantibody testing in serological evaluation of patients may delay or miss the diagnosis. A 68-year-old man presented with increasing involuntary movements consistent with faciobrachial dystonic seizures (FBDS). Initial evaluation demonstrated VGKC antibody seropositivity with leucine-rich glioma-inactivated 1 (LGI1) and contactin-associated protein-like 2 (CASPR2) seronegativity. Aggressive immunotherapy with methylprednisolone and plasmapheresis was started early in the course of his presentation. Following treatment with immunotherapy, the patient demonstrated clinical improvement. Repeat serum evaluation 4 months posthospitalisation remained seropositive for VGKC-complex antibodies, with development of LGI1 autoantibody seropositivity. VGKC-complex and LGI1 antibodies remained positive 12 months posthospitalisation. Our findings suggest that clinical symptoms can predate the detection of the antibody. We conclude that when suspicion for autoimmune encephalitis is high in the setting of VGKC autoantibody positivity, regardless of LGI1 or CASPR2 seropositivity, early immunotherapy and repeat testing should be considered.
This is an Open Access article distributed in accordance with the Creative Commons Attribution Non Commercial (CC BY-NC 4.0) license, which permits others to distribute, remix, adapt, build upon this work non-commercially, and license their derivative works on different terms, provided the original work is properly cited and the use is non-commercial. See: http://creativecommons.org/licenses/by-nc/4.0/
Statistics from Altmetric.com
Background
Antibodies directed against components of the voltage-gated potassium channel (VGKC)-complex are associated with several clinical syndromes, including limbic encephalitis, focal epilepsy, Morvan syndrome and neuromyotonia.1–4 Neurologic symptoms in patients with VKGC-complex antibodies have been described in peripheral neuropathies, myelopathy and several movement disorders.5
Testing for VGKC-complex antibodies is often performed via an immunoprecipitation assay using VGKCs solubilised from mammalian brain homogenates in digitonin and ligated with radioiodinated α-dendrotoxin (125I-α-DTX).6 Specific components of the VGKC-complex, including the leucine-rich glioma-inactivated 1 (LGI1) and contactin-associated protein-like 2 (CASPR2) antigens, coprecipitate in the immunoprecipitation assay used to detect VGKC antibodies, and can also be tested for more specifically via indirect immunofluorescence. A commercially available monospecific test using CASPR2-transfected or LGI1-transfected human embryonic kidney (HEK) cells is commonly utilised by many referral laboratories for clinical sample analysis.7 ,8 In patients who have VGKC antibodies detected, half also test positive for LGI1 or CASPR2 antibodies.9 The clinical significance of VGKC antibody positivity in the absence of LGI1 or CASPR2 antibody positivity remains an area of controversy given the variable clinical presentation of these patients and the variable response to immunotherapy.9
Here, we describe a case of autoimmune VGKC-complex encephalitis with LGI1 and CASPR2 seronegativity at initial presentation, which subsequently seroconverted to LGI1 antibody positivity several months later. Our case underscores the importance of testing for VGKC-complex antibodies, and cautions against strict reliance on only LGI1 and CASPR2 antibody testing.
Case presentation
A 68-year-old man with medical history notable for hypothyroidism, hypertension, dyslipidemia and previous cardiac arrest presented with 6 weeks of involuntary, recurrent, episodic contractions of the left face and right hand. Symptoms progressed over several weeks to involve all extremities, occurring at intervals of about 10 min when first evaluated by his local neurologist. Initial MRI brain and cervical spine with and without contrast and routine EEG were normal. Treatment with magnesium, calcium, diazepam, lorazepam, baclofen and levetiracetam were unsuccessful. He experienced mild relief with clonidine and clonazepam.
On evaluation in our clinic, examination demonstrated episodic left face tightening and grimacing along with bilateral arm flexing and hand tightening, consistent with faciobrachial dystonic seizures (FBDS). He also demonstrated fast myoclonic jerking in his right arm, right foot raising, restless right leg movement and decreased right arm swing when he walked (video 1). Montreal cognitive assessment during initial evaluation was 24. He was admitted to hospital for expedited evaluation and treatment.
Presentation at time of presentation. Clinical findings consistent with faciobrachial dystonic seizure including facial grimacing with associated bilateral are flexion with hand clenching. He also demonstrates myoclonic movement in his right arm and restless leg movement in his legs.
Investigations
Cerebrospinal fluid (CSF) had 1 white cell count/HPF, 669 red blood cells, protein 30 mg/dL, glucose 79 mg/dL, all within normal; viral infectious studies were negative. Autoimmune serological evaluation was positive for serum VGKC-complex antibody at 698 pmol/L (normal 0–31 pmol/L, Associated Regional and University Pathologists (ARUP) Laboratories). Results for serum and CSF follow-up testing of LGI1 and CASPR2 IgGs at ARUP Laboratories and Mayo Medical Laboratories (MML) were negative.
MRI brain with and without contrast repeated 1 month following initial imaging demonstrated increased right hippocampal and amygdala signal on T2/fluid-attenuated inversion recovery (FLAIR) sequence (figure 1), although imaging did not demonstrate basal ganglia T1 hyperintensity previously described in patients with FBDS.10 Continuous EEG revealed no slowing or epileptiform activity despite capturing multiple events on video recording. CT chest, abdomen, pelvis revealed no malignancy. He was diagnosed with FBDS associated with VGKC-complex antibodies, despite LGI1 and CASPR2 seronegativity.

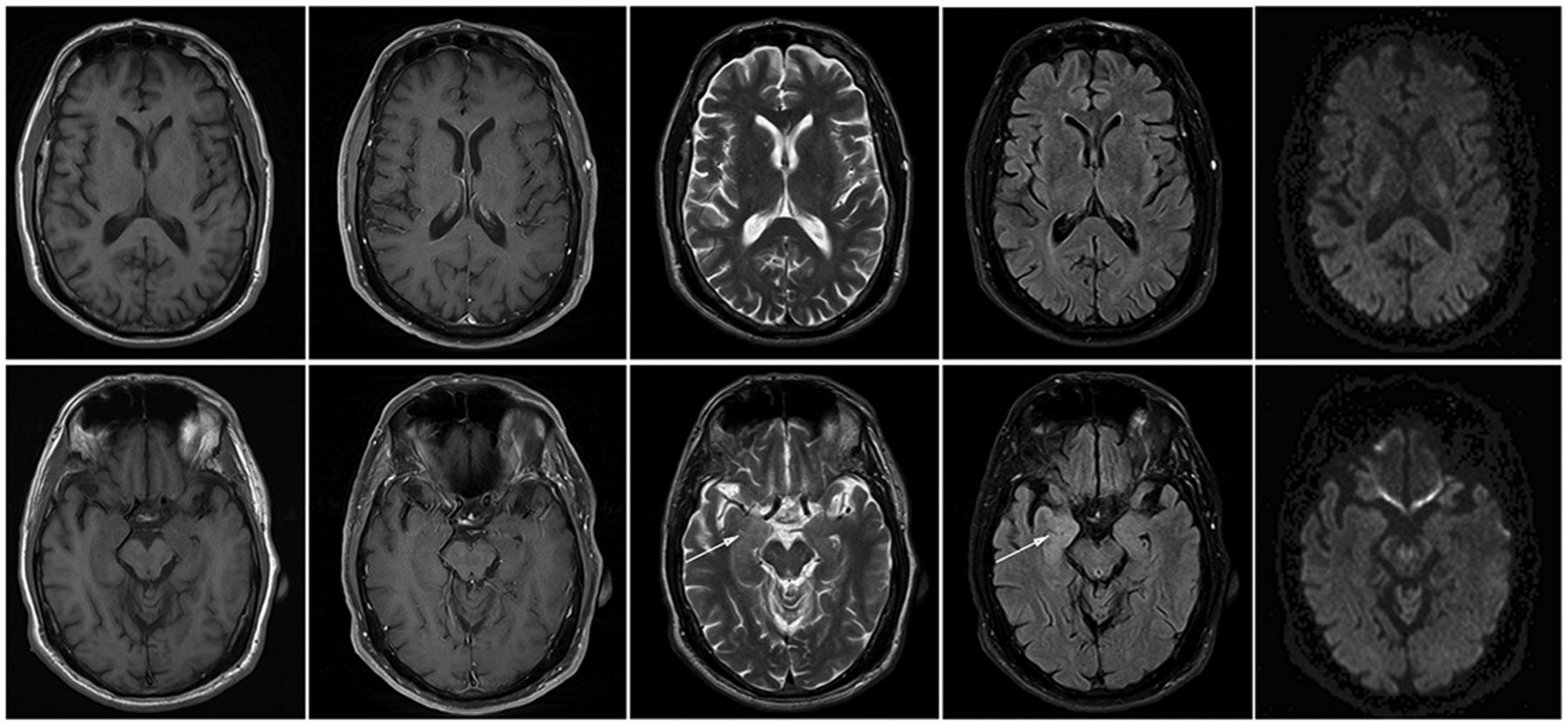
Brain MRI images at time of presentation. Images from left to right: T1w precontrast, T1w postcontrast, T2w, FLAIR, diffusion tensor imaging (DTI) trace with 20 direction B2000. Top: MRI images at the level of the basal ganglia without abnormal signal on any of the sequences. Bottom: MRI images at the level of the hippocampus demonstrate high T2/FLAIR signal in the right amygdala (arrows), but no abnormal enhancement of restricted diffusion.
Repeat MRI brain 2 months posthospitalisation revealed persistent increased hippocampal and amygdala signal on T2/FLAIR sequence. Repeat routine EEG demonstrated right frontotemporal slowing.
Repeat serum evaluation performed 4 months posthospitalisation (6 months from symptom onset) was remarkable for a positive LGI1 antibody at 1:160 end point titre (ARUP Laboratories).
Differential diagnosis
The differential diagnosis for possible autoimmune encephalitis is quite broad, including neoplasm, atypical presentation of epilepsy (causing limbic hyperintensity on MRI), and vascular aetiologies causing focal seizures (ischaemic or haemorrhagic stroke, or vascular malformations). Wernicke's encephalopathy and other metabolic derangements should also be considered, as well as toxin exposure or adverse drug reaction. Commonly encountered infectious possibilities include herpes simplex virus, varicella zoster virus, West Nile virus, enterovirus, tuberculosis and listeria. Prion disease may also present in similar fashion and has been reported to mimic autoimmune encephalitis. Finally, while unlikely in our patient, given his age of presentation, heritable disorders may present similarly to encephalitis. In regard to autoimmune or paraneoplastic aetiologies, there are multiple antibodies—known and as yet unclassified—that are associated with autoimmune encephalitis.
Treatment
He was initially treated with a 5-day course of intravenous methylprednisolone followed by a 5-day course of plasmapheresis with some improvement in seizure frequency, severity and duration. Following discharge, he was treated with additional intravenous methylprednisolone, after which mycophenolate mofetil was initiated.
Two months posthospitalisation, the patient reported persistent episodes of involuntary facial and arm movements, improved immediately after each methylprednisolone treatment. He was transitioned to oral prednisone, along with continuation of mycophenolate mofetil.
Four months posthospitalisation, he reported increased frequency of his FBDS after transition to oral steroids and if his daily sodium intake was low. Serum and urine sodium studies confirmed syndrome of inappropriate antidiuretic hormone secretion (SIADH) and he was started on daily sodium supplementation. He was started on valproate sodium and underwent a repeat 5-day course of plasmapheresis.
Outcome and follow-up
Twelve months posthospitalisation, he reported less frequent and less severe seizures with treatment with valproate sodium, mycophenolate mofetil and prednisone. A repeat serum evaluation was again notable for VGKC-complex antibody positivity at 337 pmol/L, LGI1 antibody positivity at 1:160 end point titre and negative CASPR2 antibody (ARUP Laboratories). After therapy and 1 year from symptom onset, the changes on MRI had resolved (figure 2). On last follow-up, he was essentially symptom free, on stable doses of valproate sodium and mycophenolate mofetil and completely off prednisone. Neurological examination was essentially normal with the exception of 2/3 delayed recall and a Montreal Cognitive Assessment Score of 24/30 on intial evaluation, with subsequent normalisation after treatment.
{kind=link}
{kind=link}
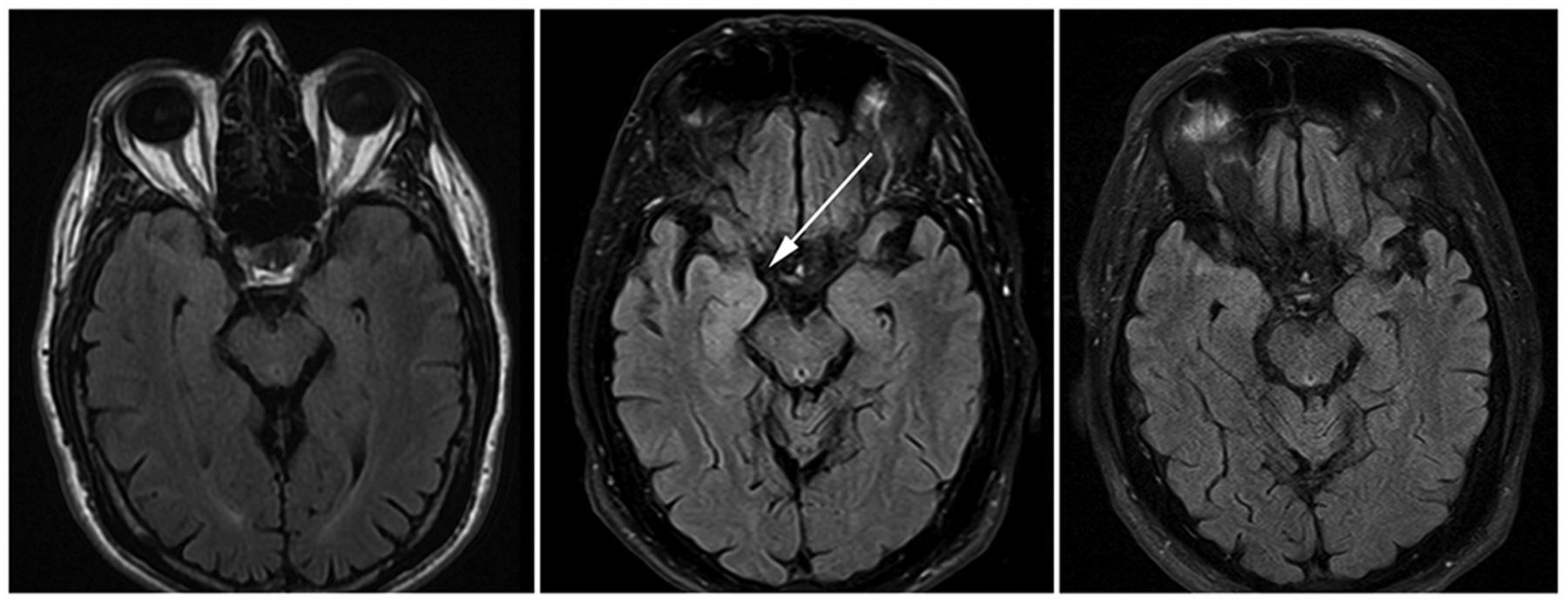
Brain MRI images from time of presentation to our clinic. Brain MRI FLAIR images from left to right: 1 month prior to evaluation at our clinic, at time of evaluation in our clinic, and 1 year from symptom onset and initial MRI. FLAIR hyperintense signal in the right amygdala (arrow) was not evident on presentation MRI, but became positive 1 month later, and decreased to normal levels 1 year from symptom onset.
Discussion
Originally, VGKC-complex antibodies were isolated from patients with neurologic syndromes including neuromyotonia, Isaacs syndrome and Morvan syndrome. The spectrum of phenotypes associated with VGKC-complex antibodies has since grown and resulted in additional research into the VGKC complex. In 2006, it was described that sera from patients with neuromyotonia, Morvan syndrome and limbic encephalitis were positive for Kv1.1, Kv1.2 or Kv1.6 antibodies by immunoprecipitation of 125I-α-DTX-labelled rabbit brain K+ channels. In cells transfected with combinations of several Kv subunits of VGKC, sera from patients with limbic encephalitis preferentially bound with Kv1.1 and sera from patients with Morvan syndrome and neuromyotonia preferentially bound to Kv1.2 or Kv1.6 channels. Only 20–38% of transfected cells were recognised by patient antibodies. This suggested the presence of antibodies with different antigenic specificity may explain the presence of different clinical phenotypes.11
In 2010, sera and CSF from 57 patients with VGKC seropositivity and limbic encephalitis were tested using HEK293 cells transfected with LGI1. All 57 patient samples demonstrated binding to LGI1. It was concluded that autoantibodies to LGI1 were associated with limbic encephalitis.7 Another study demonstrated that sera from 96 patients with VGKC autoantibodies bound to VGKC-associated proteins including CASPR2 and LGI1 rather than directly to VGKC subunits.12 These studies support the notion that VGKC autoantibodies detected by radioimmunoprecipitation assays actually bind to other synaptic or axonal proteins that coprecipitate with the solubilised VGKC proteins. Thus, it became unclear how to proceed clinically in cases where VGKC autoantibodies are detected in the absence of LGI1 or CASPR2 negativity.
A study in 2016 examined 1455 subjects tested for VGKC-complex antibodies by radioimmunoassay. Of 50 patients (age range 1–84 years) who tested positive (titre>100 pM) for VGKC-complex antibodies, 25 were LGI1 or CASPR2 seropositive. VGKC-positive patients without LGI1/CASPR2 antibodies fulfilled criteria for autoimmune inflammation in 7 of 25 (28%) cases. Of patients lacking LGI1/CASPR2 antibodies, improvement with immunotherapy was noted in 11 of 24 (46%) cases.9 This demonstrates that a subset of VGKC-positive patients will be LGI1/CASPR2 seronegative, yet still meet clinical criteria for autoimmune inflammation and respond to immunotherapy. More recently, a study of 72 double-negative VGKC complex antibodies found that 10 (14%) immunoprecipitated 125I-α-DTX itself, and 27 (38%) bound to Kv1.1/1.2/1.6 subunits and/or Kv1.2 subunits alone, leading the authors to suggest that the presence of double-negative VGKC-complex antibodies in themselves support the use of immunotherapies, but depending on clinical phenotype, should prompt further investigation for other antibodies or another immune-mediated pathology.13
In our patient, initial VGKC antibody titre was significantly elevated at 698 pmol/L. Given the subacute presentation with FBDS, encephalopathy, and right amygdala and hippocampus hyperintense T2 signal, suspicion for autoimmune encephalitis was high. Testing for LGI1 and CASPR2 antibodies, however, was negative at two national referral laboratories. Despite negative testing for these specific antibodies, empiric treatment with immunotherapy was initiated as our patient met clinical criteria for autoimmune encephalitis proposed by Graus et al. He demonstrated clear clinical effectiveness with improvement in encephalopathy and decreased frequency of seizures. One year following initial presentation and negative LGI1 testing, our patient had ongoing seizures. Repeat antibody evaluation demonstrated an LGI1 antibody titre in the serum at 1:40 and 1:160 (MML and ARUP Laboratories, respectively).
In a study of 26 patients with limbic encephalitis and VGKC antibodies detected by radioimmunoassay, 23 had LGI1 antibodies detected as well. In this group, four patients had VGKC antibody and LGI1 testing following onset of FBDS but prior to onset of encephalopathy with four positive for VGKC antibodies and three positive for LGI1 antibodies.14 This suggests antibodies are present early in the clinical course. In our case, symptoms were present for at least 6 months prior to LGI1 antibody detection. This could be indicative of assay limits, as the VGKC radioimmunoassay detects more of the complex than just the LGI1 or CASPR2 antigens. While it is unclear why the initial LGI1 assay was negative—one possible explanation is the initial antibody titre tested with the commercially available LGI1 cell-based immunofluorescence assay was too low to be visually detected, exceeding the lower limits of the assay sensitivity. However, given that the patient was symptomatic of the classic syndrome associated with LGI1 antibodies at the time of testing, this is not a trivial observation. It suggests VGKC-complex antibody testing may be a more sensitive indicator earlier in disease course in some patients—or at least that the VGKC-complex assay can be more sensitive than commercially available LGI1 cell-based assays. VGKC-complex antibody testing continues to have a role in screening, as it can at a minimum support further investigation into an immune-mediated aetiology in the appropriate clinical setting.
This case illustrates several important points that should be considered when evaluating and treating patients with suspected autoimmune encephalitis. Treatment with early immunotherapy should be considered when the index of suspicion for autoimmune encephalitis is high,15 as clinical symptoms may predate detection of a specific antibody. Repeat testing should be considered, and the presence of VGKC-complex antibodies alone in the appropriate clinical context warrants prompt and aggressive immunotherapy.
Learning points
Antibodies directed against components of the voltage-gated potassium channel (VGKC)-complex are associated with several clinical syndromes, including limbic encephalitis.
The clinical significance of VGKC antibody positivity in the absence of leucine-rich glioma-inactivated 1 (LGI1) or contactin-associated protein-like 2 (CASPR2) antibody positivity remains an area of controversy.
We describe a case of autoimmune VGKC-complex encephalitis with LGI1 and CASPR2 seronegativity at initial presentation, which was empirically treated with immunotherapy due to clinical presentation consistent with autoimmune encephalitis. He subsequently seroconverted to LGI1 antibody positivity several months later.
Treatment with early immunotherapy should be considered when the index of suspicion for autoimmune encephalitis is high, regardless of LGI or CASPR2 status, as clinical symptoms may predate detection of a specific antibody.
Acknowledgments
The authors are grateful for the support of the Transverse Myelitis Association (TMA) and Barbara Gural-Steinmetz. We also appreciate the assistance of the Neuroimmunology Laboratory at Mayo Medical Laboratories.
References
Footnotes
Contributors MS evaluated the patient, analysed and interpreted the data, and drafted and revised the manuscript. JG evaluated the patient, analysed and interpreted the data, and drafted and revised the manuscript. SMcN analysed and interpreted the data, and created imaging figures. SC clinically evaluated the patient, analysed and interpreted the data, and revised the manuscript.
Competing interests None declared.
Patient consent Obtained.
Provenance and peer review Not commissioned; externally peer reviewed.