Article Text

Statistics from Altmetric.com
Description
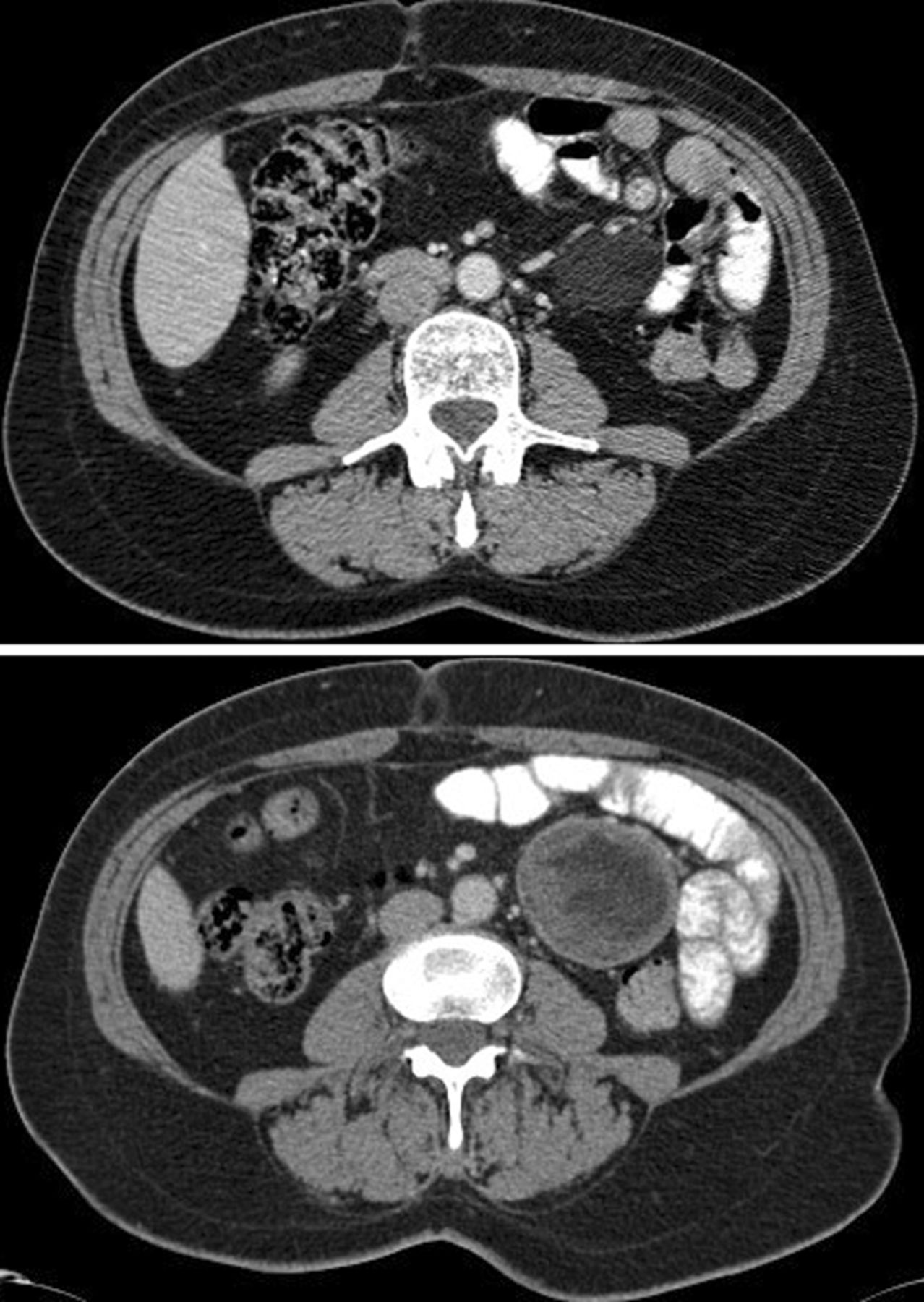
An asymptomatic 56-year-old woman with a history of multiple basal cell carcinomas (BCCs) and odontogenic keratocysts was referred to the metabolic clinic for assessment of bone density. On examination, there were no plantar or palmar pits, facial abnormalities or macrocephaly and the family history was also unremarkable. Osteoporosis was excluded, but plain films showed multiple cysts throughout the long bones (figure 1) and subsequent abdominal CT revealed multiple large mesenteric cysts (figure 2). Multiplex ligation-dependent probe amplification analysis confirmed a heterozygous pathogenic deletion in patched 1, consistent with a diagnosis of Gorlin syndrome.

Plain X-rays showing diffuse long bone cysts.
{kind=link}
{kind=link}
CT scan showing a large mesenteric cyst.
Gorlin syndrome, or naevoid BCC syndrome, is a rare autosomal dominant condition of variable expression, which has an estimated prevalence of 1 in 164 000 in the UK.1
Gorlin syndrome can present with a wide range of multisystem manifestations. Clinical features include the early onset BCCs, macrocephaly, palmar or plantar pits and coarse facies. Common radiological findings include benign keratocystic odontogenic tumours of the jaw, rib abnormalities and early calcification of the falx cerebri.2 The radiological features in this case have been described less frequently, with polyostotic bone cysts being present in less than 50% and lymphomesenteric cysts in less than 5% of individuals with Gorlin syndrome.3
Examples of common radiological findings have been widely published, but these striking images demonstrating large bone cysts with diffuse axial involvement and lymphomesenteric cysts are unusual even in this rare disease.
Learning points
-
Gorlin syndrome is a rare condition of great clinical importance due to the predisposition to benign and malignant tumours.
-
The commonest features are early onset basal cell carcinomas, odontogenic keratocysts and early calcification of the falx cerebri.
-
Although diagnosis is frequently established using clinical diagnostic criteria, PTCH1 is the only gene in which mutations are known to cause Gorlin syndrome.
Acknowledgments
The authors would like to thank Dr Bill Newman for providing information on genetic testing and results.
Footnotes
-
Contributors JEH and JS participated in the writing of the manuscript. HS contributed to identifying the case and to overseeing the overall content. RWW was involved in the identification and formatting of the images.
-
Competing interests None.
-
Patient consent Obtained.
-
Provenance and peer review Not commissioned; externally peer reviewed.