Article Text

Statistics from Altmetric.com
Description
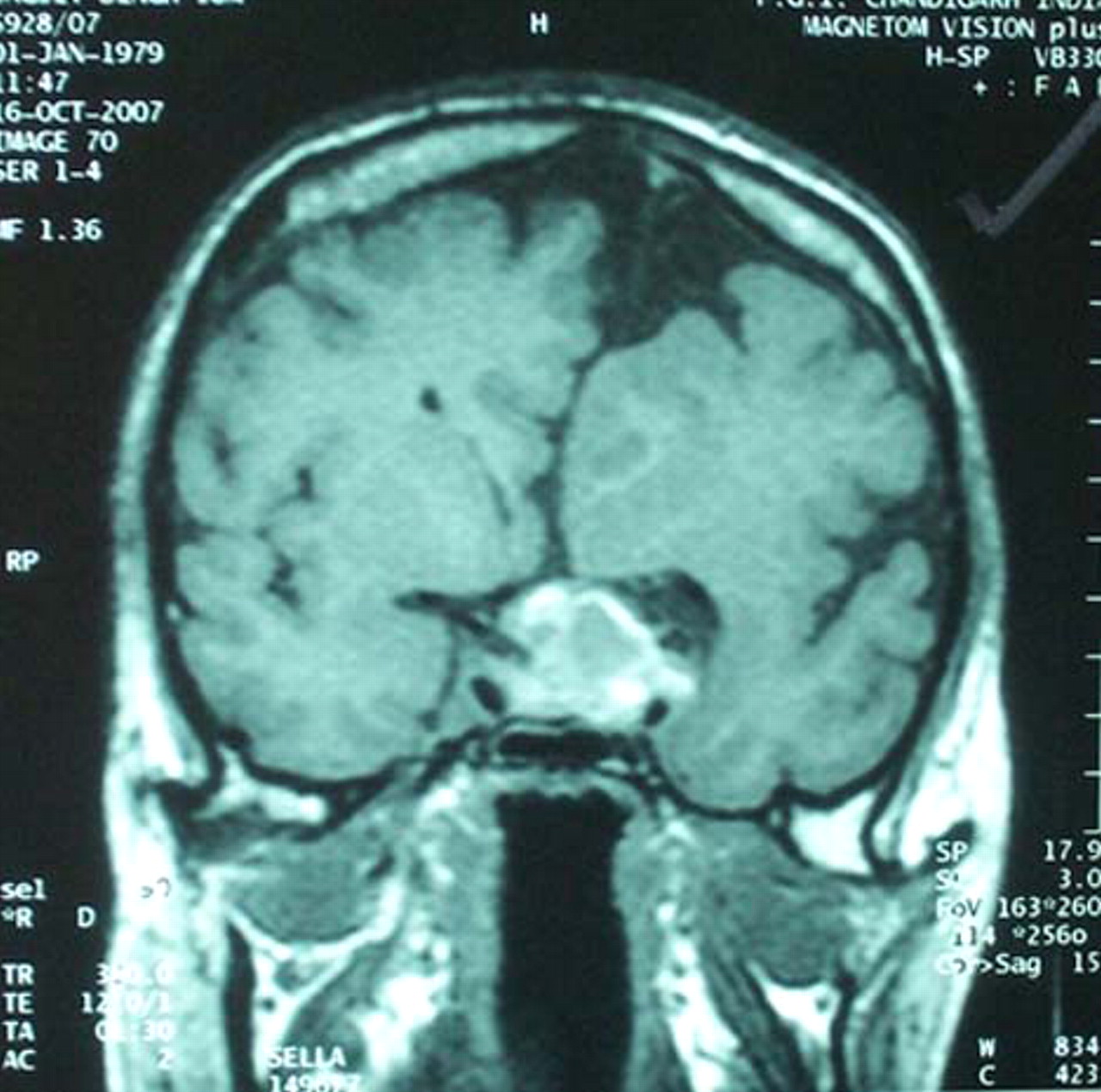
Midline developmental abnormalities are usually associated with isolated growth hormone deficiency or multiple pituitary hormone deficiencies due to pituitary transcription factor defects.1 We describe a case with midline defects presenting as corpus callosum agenesis and acromegaly. A 39-year-old male presented with congestive cardiac failure with overt clinical features of acromegaly. He was a graduate, was mentally normal and had no visible morphological abnormality. On evaluation, serum growth hormone after 75 g of anhydrous glucose load was non-suppressible (12 μg/l, normal <1 μg/l) and insulin-like growth factor-1 was elevated (900 μg/l, normal 114–49 μg/l). He was hypothyroid, hypocortisolic and hypogonadal and was on replacement therapies. MRI of the head showed a pituitary macroadenoma (2×1.5×1 cm) with left parasellar extension and subacute apoplexy and incidental findings of complete agenesis of corpus callosum (ACC) and right cerebral hemiatrophy (figure 1 and 2). His karyotype was 46, XY with no chromosomal abnormalities. The patient underwent transsphenoidal surgery and was cured.

MRI sagittal section of the brain showing complete corpus callosum agenesis.
{kind=link}
{kind=link}
Coronal section of MRI of the brain and sella showing a pituitary macroadenoma with left cavernous sinus extension and subacute apoplexy with corpus callosum agenesis and right cerebral hemiatrophy.
The corpus callosum is a band of white matter structure connecting the cerebral hemispheres medially. It develops from the lamina reuniens in the telencephalon. ACC is an anomaly that may occur in isolation or in association with Dandy–Walker cyst, congenital hydrocephalus, Arnold–Chiari malformation, holoporencephaly, polymicrogyria and sometimes with chromosomal anomalies such as trisomy 18, 13 and 8.2 Signs and symptoms of ACC vary from asymptomatic presentation as seen in our case, to vision impairment, hypotonia, delayed motor milestones, learning disabilities and, rarely, seizures and spasticity.3 This seems to be a chance association and extremely rare in acromegaly.
Footnotes
-
Competing interests None.
-
Patient consent Obtained.